基因家族分析从零到有(研究对象确定+基因家族鉴定+基本信息网站+结构分析+共线性分析+实验验证简述)
基因家族分析从零到有,本文将详细介绍每一步该做的准备。
一、介绍
在开始之前,我们需要知道,基因家族是什么?以及为什么要做基因家族分析?
基因家族(gene family):来源于同一个祖先,由一个基因通过基因复制或者加倍而产生两个或更多的拷贝而构成的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物。

202303图更正:同源信息如下图:A1A2为直系同源,A1B1为旁系同源。祖先基因拷贝变异形成A、B,进一步拷贝变异生成A1|A2|B1|B2|B3。A1A2为直系同源,结构相似功能却不一定;A1B1为旁系同源,功能相似结构不相似。
graph
a(祖先基因)-->A
a-->B
A-->A1
A-->A2
B-->B1
B-->B2
B-->B3
基因家族的遗传进化:
- 协同进化( Concerted Evolution ) :两个相互作用的物种在进化过程中发展的相互适应的共同进化。一个物种由于另一物种影响而发生遗传进化的进化类型。例如植物由于病原菌所施加的压力而与抗性基因表现出协同进化关系。
- 无功能化( Degeneration ): 由于有害突变的(非同义突变,可变剪切突变等等)不断积累,导致基因功能丧失,例如一些假基因。
- 新功能化( Subfunctionalization ): 基因在复制的过程中通过突变,遗传漂变等等使得一些基因有了新的功能
基因家族分析意义:
- 快速在目标物种中确定与目标性状有关的基因家族成员将有助于缩小研究范围
- 确定当前物种是否有特有的基因家族分支,这个可能对应了该物种特有的分子生物学过程
- 确定当前物种是否在某个特定的基因家族分支得到扩展,这个也可能对应了该物种强化的分子生物学功能,如桉树的MYB
- 了解基因的进化历史,高档次的基因家族分析,会涉及到进化尺度上的基因家族分析,厘清其进化历史,可以为遗传操作提供参考
- 补充注释信息
二、分析前准备
1)确定要研究的基因家族
与课题相关的基因家族
例如假如你们实验室是专门做某些基因的研究,对该基因家族有一定的了解,例如抗性基因,你就可以专门深挖该基因家族的研究
研究的热点
研究热点基因家族在别人没有研究的植物中的作用。如:
- WRKY参与植物生长发育、形态建成与抗病
- NBS-LRR植物中最大抗病基因家族
- MADS-box: 植物中最大的转录因子家族之一,广泛存在于真核生物中,参与花器官发育,开花时间调节
- HSP70高度保守的分子伴侣蛋白,热激活蛋白
新基因家族
找到没人没有研究过的基因家族进行研究,难度较大
在确定好研究对象后,可以通过Google或者baidu对该基因家族进行搜索,确认在你需要研究的作物中,该基因家族还没有被研究过。
2)了解你研究的基因家族的特征
参考收录了基因家族特征的网站
如 TAIR(拟南芥作为一个模式植物所被研究的基因家族),TIGR(玉米)或 PlantTFdb(主要针对转录因子),并进行预测和总结。
查找相关文献
并不是所有的基因家族都被收录到网站中,在这种情况下,我们可以通过查找与该基因家族的文献进行阅读,一般在这些文章中,都会对该基因家族进行总结规律。阅读相关文章也是很重要的,特别是同一基因家族在不同作物上的研究,你不但可以了解到其基础特征,还能通过文章了解他们所做的分析(包括方法还有参数等),对你后续的分析还有文章的写作都会有很大帮助。
方法:流程,pfamd编号
参数:保守的结构域(在哪段),几个分支,保守的aa位点,长度波动,蛋白序列
3)数据下载
在了解相关信息之后,我们将所需的数据下载下来
下载研究物种的基因组序列文件
.fa和注释文件.gtf/.gff3。常用的植物数据库,例如ensembl plant 和 Phytozome 里面囊括了大部分的植物的基因组文件。也可以从文献中看前人使用的是哪里的数据,下载不了可以试着联系原作者
下载所感兴趣的基因家族隐马可夫模型
.hmm文件下载参考基因组的基因序列
.fa文章的附件或tair、UniProtKB/Swiss-Prot等网站中搜索得到,复制然后保存为fasta格式
知道access可以通过这个网址批量下载https://www.ncbi.nlm.nih.gov/sites/batchentrez
关于UniProtKB/Swiss-Prot和UniProtKB/TrEMBL
- Swiss-Prot:高质量的、手工注释的、非冗余的数据集
- TrEMBL:电脑自动注释的
三、基因家族成员的鉴定
获取候选基因家族成员有两个策略。 一是用下载好的hmm模型或自己构建的HMM moedl模型作为query,来搜索马铃薯蛋白序列数据。在这里就需要用到hmmer子程序
hmmsearch。 二是用已发表的确定的MADS-box序列为种子序列(query),直接本地blast获取同源基因。
1)确定基因家族
1. 比对
用下载得到的已经确认的基因家族基因作为参考,进行序列比对检索
blast
# 建库
makeblastdb -in protein.faa -dbtype prot -out p_Blast -hash_index -parse_seqids
# 比对
blastp -query WRKY_eggplant.fa -db p_Blast -evalue 1e-5 -outfmt 6 -out m2e_WRKY.blast -parse_seqids
参数介绍:
# 建库
makeblastdb -in input_file -dbtype prot -parse_seqids -out database_name -logfile File_Name
-in:输入数据
-dbtype:序列类型,nucl为核酸,prot为蛋白
-parse_seqids:推荐加上
-out:输出建立的数据库
-logfile:日志文件,如果没有默认输出到屏幕# 比对
blastp -query seq.fasta -out seq.blast -db dbname -outfmt 6 -evalue 1e-5
blastn,blastp:序列类型
-query: 输入文件路径及文件名
-out:输出文件路径及文件名
-db:格式化了的数据库路径及数据库名,上一步建成的
-outfmt:输出文件格式,总共有12种格式,6是tabular格式对应之前BLAST的m8格式
-evalue:设置输出结果的e-value值
-num_threads:线程
或者使用diamond,会更快
# 建库
# 会将fasta格式的蛋白序列创建成为后缀为.dmcd的数据库文件
diamond makedb --in protein.faa -d p_diamond
# 比对
diamond blastp --query WRKY_eggplant.fa -d p_diamond -o m2e_diamond.blastp -e0.00005
参数介绍:
# 建库
diamond makedb --in --db``–in
:输入文件–db:输出的数据库前缀,或–nr`# 比对
diamond blastx --db -q reads.fna -o dna_matches_fmt6.txt比对蛋白:
diamond blastx比对dna序列:
diamond blastp
--query/-q:输入检索序列,下载的已经确定的基因序列
--out/-o:输出文件,默认以–outfmt 6输出结果和BLAST+的–outfmt 6结果一致。
-p线程
blast结果说明???
https://www.plob.org/article/641.html
主要是第三列pident(percentage of identical matches)和第四列length(alignment length),去冗余
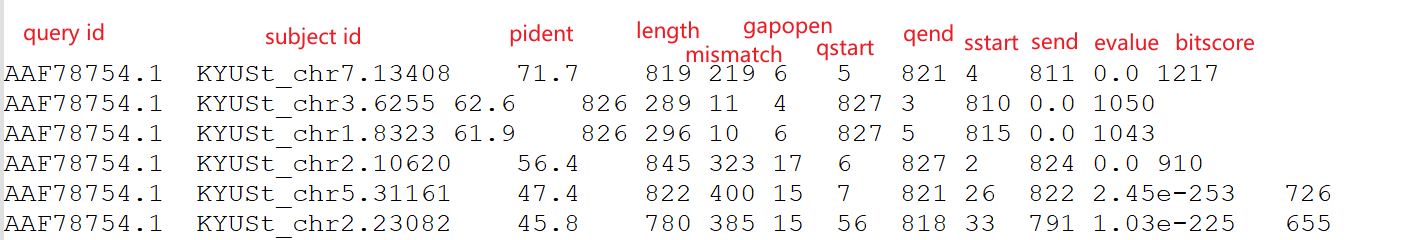
2. hmmer
先在全基因组的范围内使用
hmmersearch和基因家族的隐马可夫模型.hmm进行基因家族的进行初步搜索,接着把质量比较高的基因家族候选基因筛选出来E-value < 1 × 10−20。没有模型的使用clustalw2对高质量的序列进行多序列比对,多序列比对后,对这些置信的序列进行隐马可夫模型的构建(使用hmmbuild),最后使用该新建的隐马可夫模型,进一步筛选完整的NSB基因家族序列(需再次过滤,找到基因家族的成员数量一般比第一步初步筛选的多)
在mega里对参考基因组进行clustalw比对,保存为.fas,然后利用hmmbuild建库,hmmsearch搜索基因家族
这是没有隐马可夫的情况下。如果有,可以直接
hmmsearch,不用建库
# 以下是步骤
# 解压
gzip -d .gz
# 建库
hmmbuild WRKY.hmm WRKY_eggplant.fas
# 搜索基因家族
hmmsearch WRKY.hmm protein.faa >WRKY_hmm.out
3.验证
excel里合并上述两种方法的gene id,并删除重复值
找出两种方法共有的基因家族,这样结果就比较置信了。
通过
tbtools的gene simplify和fasta extract,导出为.fa,获得初步筛选的蛋白序列fasta信息基因组文件里面的gene id都很多描述,简化之后方便比对;
fasta extract是通过gene id得到蛋白序列如果只用了blast,需要借助
NCBI-CDD或者pfam或者smart按照关联结构域对家族蛋白进行进一步筛选。网址:NCBI CD-Search tool, Pfam ,InterProScan sequence search。
在查看保守结构域后,如果该区域含有基因家族所对应的保守结构域,例如LRR区域等,该蛋白质序列可以保留进行后续的分析。如果在该区域没有找到对应的保守区域,为了分析的严谨性,需进行进一步的排查来确定是否要去掉该序列。这种情况一般分为两种情况,第一种就是注释无误,该序列确实丢失了对应的保守结构域,需要去掉。第二种情况就是注释有误,该序列的结构域可能没有被完整的保留下来,这种情况应该截取该序列的上下游重新注释分析。
四、基因家族成员基本信息分析
在得到基因家族成员信息后,通常会根据其蛋白序列预测其基本信息,并汇总成一个表,包含gene ID|subfamily|CDS length|protein length|Molecular weight|等电点|CDS sequence|protein sequence|
蛋白物理化学性质分析
- 等电点和分子量,可以批量分析:https://web.expasy.org/compute_pi/
- 各种理化性质,需要一个个输入序列:https://web.expasy.org/protparam/
- 难看出来,哪个数字大就定位到哪:http://linux1.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc
亚细胞定位
植物:http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/#
各物种:http://cello.life.nctu.edu.tw/
跨膜结构域预测 https://services.healthtech.dtu.dk/service.php?SignalP
信号肽预测 https://services.healthtech.dtu.dk/service.php?SignalP
使用SignalP 注释蛋白序列是否含有信号肽结构,使用TMHMM注释蛋白序列是否含有跨膜结构,最终筛选出含有信号肽结构并且不含跨膜结构的蛋白为分泌蛋白。
靶向信号
TBtools通过调用脚本,批量计算理化性质
五、构建进化树
- 使用
mega对这一组蛋白序列进行clustalw多序列比对 - 构建系统发育树,构树方法有邻接法(Neighbor-Joining, NJ),最大似然法(Maximum likelihood,ML),最大简约法(Maximum parsimony,MP)和贝叶斯法(Bayesian inference, BI)不同方法各有优劣。
- 美化。在获得进化树之后可以使用在线工具
EvolView对进化树进行美化,添加一些参数使其展示的内容更加丰富。也可以导入R中利用ggtree进行美化
六、保守domain、motif、基因结构分析
Motif是位于二级和三级结构之间的层次,Motif的层次接近二级结构。 Domain是位于二级和三级结构之间的层次,Domain的层次接近三级结构
区分家族成员时,motif分析可以看出区别,但要知道区别是否有独立功能需要看domain
domain
上传蛋白序列到:SMART,NCBI CD-Search tool, Pfam ,InterProScan sequence search,其中一个就可以
tbtools上安装插件:batch smart,可以批量下载smart比对信息
motif
上传蛋白序列到:MEME
参数设置如下图,运行后保存.xml文件,可导入tbtools分析

mod zoops motif的分布类型
- oops 每个功能域在每一段序列中都会出现一次,而且只出现一次。这种模式是运算速度最快,而且最为敏感的。但是如果并不是每个序列都包含功能域,那就可能会有不正确的结果。
- zoops 每个功能域在每一段序列中至多只出现一次,可能不出现。这种模式运算速度较快,敏感性稍弱。
- anr 每个功能域在每一段序列中出现的次数不定。这种模式运算速度最慢,可能会多花十倍以上的时间。但是对于功能分布的情况完全未知的情况下,这一参数可能会有帮助
Select the number of motifs
决定在这一组多条序列中,将被挖掘出的结构域(motif)的种类数量。默认值是3,这意味着在这一组序列中,发现的motif的数量最多3个。但有时候,我们无法预先了解这组序列实际上的结构域的数量,那么可以先填写一份较大的数值。例如10。在完成分析后,再查看分析结果中结构域的显著性。例如,如果结果中保守性达到显著水平(P<0.05)的结构域数量是6。那么,则可以将最初的参数从“10”修改为“6”,然后重新提交数据分析一次。
基因结构(外显子/内含子)
以
TBtools为例,首先根据基因家族id提取对应gff文件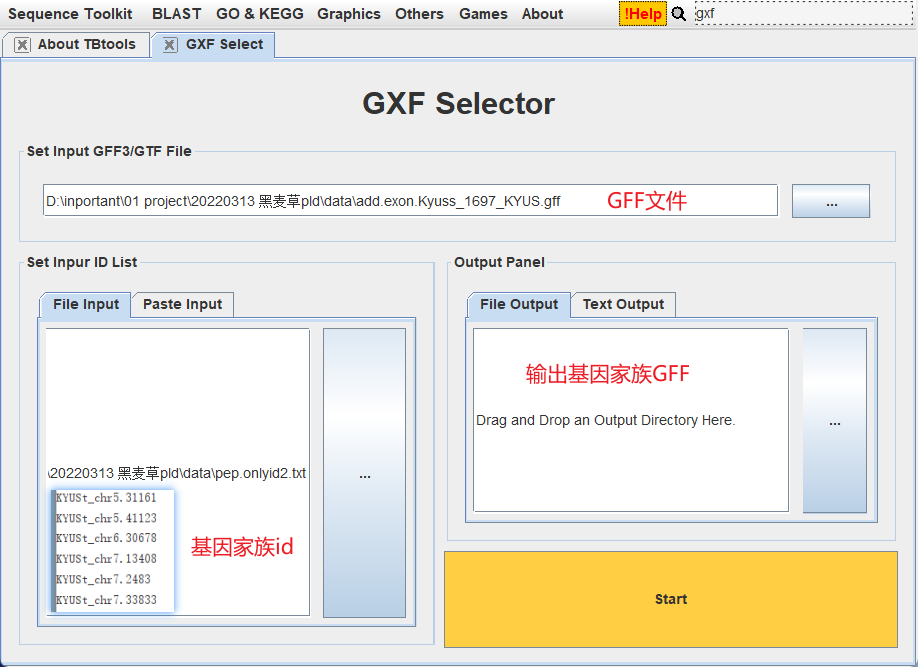
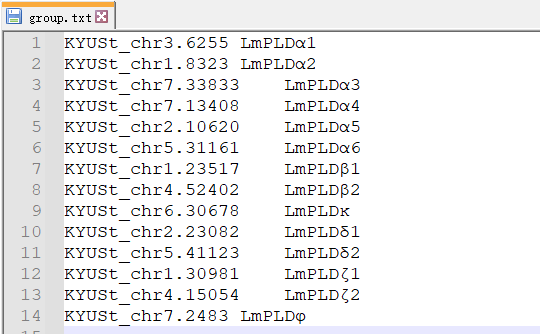
使用
Gene Structure Shower模块,将基因家族成员的id以及基因组注释gtf/gff3文件导入TBtools,就可以得到基因结构图注意检查id与注释文件中是否对应
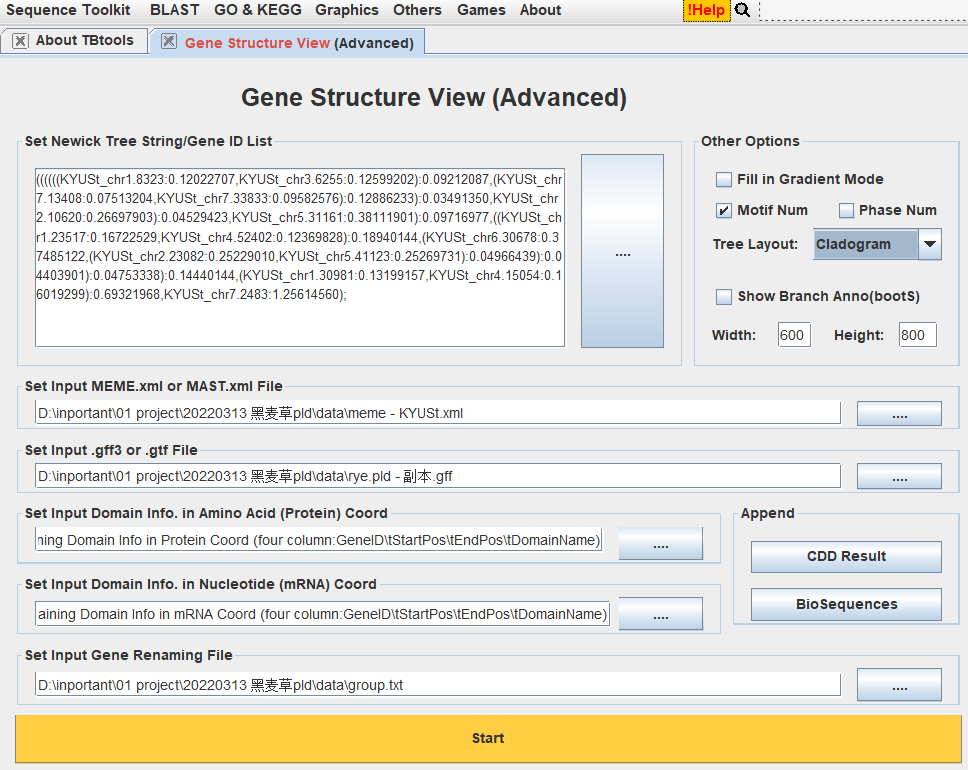
七、染色体定位/染色体共线性分析
染色体定位
根据id和注释信息即可得到,可利用tbtools
确定好要表现的染色体,一般只选择Chr,拼接的如果没有基因注释上去就不用了
染色体共线性分析
下载数据
- 所需基因组的gff文件
- 所需基因组的pep或者cds序列
MCScanX
# 解压缩
gunzip *.gz
# 1.比对
## 合并
cat Kyuss_1697_KYUS_proteins.fa Arabidopsis_thaliana.TAIR10.pep.all.fa Oryza_sativa.IRGSP-1.0.pep.all.fa > all.fa
## 建库
diamond makedb --in all.fa -d all.diamond
## 蛋白比对
diamond blastp --query all.fa -d all.diamond -o all.blastp -e0.00005
# 2.调整gff格式
## 删除前面的##说明
sed -i '1,69d' Oryza_sativa.IRGSP-1.0.52.gff3
sed -i '1,13d' Arabidopsis_thaliana.TAIR10.52.gff3
## 调整
### 只取含有基因的列
cat Arabidopsis_thaliana.TAIR10.52.gff3|awk '$3=="gene"{print $0}'| sed 's/=/\t/' | sed 's/;/\t/' | awk 'BEGIN{OFS="\t"}{print $1,$10,$4,$5}'>ara.gff
cat Oryza_sativa.IRGSP-1.0.52.gff3|awk '$3=="gene"{print $0}'| sed 's/=/\t/' | sed 's/;/\t/' | awk 'BEGIN{OFS="\t"}{print $1,$10,$4,$5}'>ory.gff
cat add.exon.Kyuss_1697_KYUS.gff|awk '$3=="gene"{print $0}'| sed 's/=/\t/' | sed 's/;/\t/' | awk 'BEGIN{OFS="\t"}{print $1,$10,$4,$5}'>rye.gff
## 合并
cat rye.gff ara.gff ory.gff > all.gff
# 3.运行MCScanX
MCScanX all
# 画图
## 定义一下输入环境,因为MCScanX的java包不知道怎么设置到PATH里
inputdir=/home/leixingyi/project/genefamily_ryegrass/mcscanx/
outdir=/home/leixingyi/project/genefamily_ryegrass/mcscanx/
index=~/applications/mcscanx/MCScanX-master/downstream_analyses/
## dot图
### 配置数据
cd $index
vi dot.ctl
800 #x
800 #y
800 #z
1,10,11,12,2,3,4,5,6,7,8,9 #rye
chr1,chr2,chr3,chr4,chr5,chr6,chr7 #ara
scaffold_1242,scaffold_1259,scaffold_1700,scaffold_1854,scaffold_2697,scaffold_2787,scaffold_3611,scaffold_6468,scaffold_719,scaffold_869 #ory
### 画图
java dot_plotter -g ${inputdir}all.gff -s ${inpudir}all.collinearity -c dot.ctl -o ${outdir}dot.PNG ##空空如也
## 不干了,直接介绍一下四种出图
java dot_plotter -g at_vv.gff -s at_vv.collinearity -c dot.ctl -o dot.PNG #dot
java dual_synteny_plotter -g at_vv.gff -s at_vv.collinearity -c dual_synteny.ctl -o dual_synteny.PNG #直线
java circle_plotter -g at_vv.gff -s at_vv.collinearity -c circle.ctl -o circle.PNG #圈圈
java bar_plotter -g at_vv.gff -s at_vv.collinearity -c bar.ctl -o bar.PNG #直方
sed不能全局删除吗,好像只能删第一个匹配项这里提取类型为gene的时候,如果是第一列可以直接
grep '^gene'
>>追加,>新建
MCScanX(python)
运行
## 修改格式 --type:gff文件中第三列 --key:type对应的第九列信息前缀,.gz可以直接输入
python -m jcvi.formats.gff bed --type=mRNA --key= Name Arabidopsis_thaliana.TAIR10.52.gff3 -o ara.bed
python -m jcvi.formats.gff bed --type=gene --key=ID Oryza_sativa.IRGSP-1.0.52.gff3 -o ory.bed
python -m jcvi.formats.gff bed --type=gene --key=ID add.exon.Kyuss_1697_KYUS.gff -o rye.bed
python -m jcvi.formats.fasta format Kyuss_1697_KYUS_proteins.fa rey.pep # ...
## 去重 获取最长转录本
python -m jcvi.formats.bed uniq ara.bed #...
## 共线性分析 --cscore=.99这个选项能够有效地筛选出best hit,建议加上,--no_strip_names是必加的参数(如果.cds和.bed都是按照上述步骤修改对应格式的话)--dbtype prot蛋白质选项
python -m jcvi.compara.catalog ortholog ara ory --cscore=.99 --no_strip_names
python -m jcvi.compara.catalog ortholog ara ory --dbtype prot --cscore=.99 --no_strip_names
## 如果遇到报错,多半是要安装python包,更新Latex
###尝试2
# 安装
conda install last
# 下载数据
python -m jcvi.apps.fetch phytozome #需要自己在phytozome注册一个账号,几分钟应该就能注册好
python -m jcvi.apps.fetch phytozome Athaliana
python -m jcvi.apps.fetch phytozome OsativaKitaake
# 修改格式
python -m jcvi.formats.gff bed --type=mRNA --key=Name OsativaKitaake_499_v3.1.gene.gff3 -o osa.bed
python -m jcvi.formats.gff bed --type=mRNA --key=Name Athaliana_167_TAIR10.gene.gff3.gz -o ara.bed
python -m jcvi.formats.fasta format OsativaKitaake_499_v3.1.cds.fa osa.cds
python -m jcvi.formats.fasta format Athaliana_167_TAIR10.cds.fa.gz ara.cds
## 去重 获取最长转录本
python -m jcvi.formats.bed uniq osa.bed
## 共线性分析
python -m jcvi.compara.catalog ortholog ara osa --cscore=.99 --no_strip_names
共线性分析结果:
ara.osa.anchors:共线性block区块(高质量)ara.osa.last:原始的last输出ara.osa.last.filtered:过滤后的last输出ara.osa.pdf:两物种间的共线性点图在这里找到确定名字
https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=PhytozomeV12
s
热知识:安装cufflinks,可以直接根据gff文件,从基因组.fa文件中直接调取cds,和pep序列
# 提取cds gffread in.gff3 -g ref.fa -x cds.fa # 提取pep gffread in.gff3 -g ref.fa -y pep.fa如果遇到了报错,请你
conda install -c conda-forge jupyter_latex_envs,再不行就是cds和bed的名字对不上,再不行就是各种软件没装上,可以在报错信息里找
然后就是dvipng
sudo apt-get update -y sudo apt-get install -y dvipng
IndexError: list index out of range:bed里按染色体顺序排序;检查是否有空行
ValueError: could not convert string to float # 输入数据格式是否正确
ZeroDivisionError: float division by zero # 检查bed与seqid是否一致,区分大小写,染色体编号要带数字,seqid与layout的顺序要一直
参考:https://blog.csdn.net/u012110870/article/details/102804593
生成.simple文件
python -m jcvi.compara.synteny screen --minsize=3 --minspan=10 --simple grape.peach.anchors grape.peach.anchors.new–minspan是规定syntenic region的总长度(i.e. 基因个数)最低阈值,默认是0,
–minsize是规定syntenic region的anchors总个数(i.e.有共线性关系的基因对数目)最低阈值,默认是0,这里注意概念不要混淆,span长度为10个基因并不代表10个基因全都是检测出的高质量anchor(只是人为规定的syntenic region的一个筛选标准而已,具有主观性和物种特异型,自己视情况给定),size才是syntenic region中高质量anchor的(最小)数目,也是自己视情况给定
可视化
# 首先生成.simple文件
python -m jcvi.compara.synteny screen --minspan=30 --simple ara.osa.anchors ara.osa.anchors.new
# 编辑配置文件seqids 和layout
##查看染色体体号
cat osa.bed | awk '{print $1}'|sort -u
cat add.exon.Kyuss_1697_KYUS.gff | awk '{print $1}'|sort|uniq -c #显示重复次数
##按染色体排序
sort -t ' ' -k1n,1 osa.bed>>osa.bed
sort -t ' ' -k1n,1 ara.bed>>ara.bed
##设置需要展示染色体号 chrM和MT都代表线粒体
vi seqids
Chr1,Chr2,Chr3,Chr4,Chr5 #ara
Chr1,Chr2,Chr3,Chr4,Chr5,Chr6,Chr7,Chr8,Chr9,Chr10,Chr11,Chr12 #osa
##设置颜色,长宽等
vi layout
## y, xstart, xend, rotation, color, label, va, bed
.6, .1, .8, 0, red, ara, top, ara.bed
.4, .1, .8, 0, blue, osa, top, osa.bed
## edges
e, 0, 1, ara.osa.anchors.simple
# 运行代码,共线性分析
python -m jcvi.graphics.karyotype seqids layout
关于layout设置,不同形状
两物种平行
#start, xend, rotation, color, label, va, bed .6, .1, .8, 0, , Rye, top, rye.bed .4, .1, .8, 0, , Rye, top, rye.bed # edges e, 0, 1, rye.rye.anchors.simple三物种平行
## y, xstart, xend, rotation, color, label, va, bed .7, .2, .4, 45, ,Ara, top, ara.bed .5, .2, .8, 0, , Rye, top, rye.bed .7, .4, .8, -45, , Osa, bottom, osa.bed ## edges e, 0, 1,rye.ara.anchors.simple e, 1, 2, rye.osa.anchors.simple三物种三角形
## y, xstart, xend, rotation, color, label, va, bed .5, 0.025, 0.625, 60, , Ara, top, ara.bed .2, 0.2, .8, 0, , Rye, top, rye.bed .5, 0.375, 0.975, -60, , Osa, bottom, osa.bed ## edges e, 0, 1,rye.ara.anchors.simple e, 1, 2, rye.osa.anchors.simple e, 0, 2, ara.osa.anchors.simple
#simple
--minspan:规定syntenic region的总长度(基因个数)最低阈值,默认是0
--minsize是规定syntenic region的anchors总个数(有共线性关系的基因对数目)最低阈值,默认是0这里注意概念不要混淆,span长度为10个基因并不代表10个基因全都是检测出的高质量anchor(只是人为规定的syntenic region的一个筛选标准而已,具有主观性和物种特异型,自己视情况给定)size才是syntenic region中高质量anchor的(最小)数目,也是自己视情况给定
#layout
两个物种染色体的位置在x轴y轴什么位置(如y轴的0.6 0.4,如果是0.5 0.5就重叠了
注意, ##edges下的每一行开头都不能有空格,rotation就是旋转角度,0就代表水平
-k命令标准输入需要指定开始和末尾sort -t ' ' -k1,1 -k2n,2 data其中 -t ’ ’ 指定使用空格分列
-k1,1 指定以第一列为关键字排序
-k2n,2 指定以第二列为关键字做数据排序
提问:sort -n 和 -u并用时为什么会只出一个结果
突出显示
#Note: 注释文件的gff 与 gtf格式互相转换–使用gffread
gff to gtf
gffread anno.gff -T -o anno.gtf
6)计算ka/ks值
KaKs_Calculator 2.0 计算基因家族中每个重复基因的非同义替换(ka)和同义替换(ks)
- Ka=发生非同义替换的SNP数/非同义替换位点数
- Ks=发生同义替换的SNP数/同义替换位点数
ka/ks
- Ka/Ks=1:假设这个基因没有受到自然选择压力,那么根据中性选择理论,非同义替换率和同义替换率应该是相同的。
- Ks/Ks«1:但实际情况下,这个比值都是远小于1的,因为一般非同义替换带来的都是有害的性状,只有极少数情况下会造成进化上的优势。
- Ka»Ks或者Ka/Ks » 1,基因受正选择(positive selection)
tbtools计算,可参考这篇
六、实验验证
cDNA提取
RNA 提取试剂盒提取RNA,反转录试剂盒进行 cDNA 第一链的合成,PCR扩增
qRT-PCR 分析
primer5.0 设计引物, PCR 进行特异性验证,验证特异性符合实验要求后进行实时定量 PCR反应
特定基因克隆
- 提取DNA并反转录为cDNA,PCR扩增
- 使用试剂盒对PCR 产物切胶回收
- 基因T载体连接转化
基因过表达载体的构建
- 试剂盒提取质粒
- 质粒酶切(琼脂糖凝胶电泳鉴定)及连接
- 将重组质粒转化大肠杆菌,利用含有 Kan 抗生素的 LB 固体培养基进行筛选并测序
基因过表达植株
- 种子消毒培养
- 侵染植株:将含有重组质粒的农杆菌活化,取 OD600 值在 1.8 左右的菌液,离心去上清后用蔗糖溶液重悬。将未完全开放的拟南芥花序(不同植物可能不一样)淹没于侵染液中 30-40 s。侵染后的植株继续培养并收获 T0 代种子。
- 转基因植株的筛选:T0 代种子消毒后,在含有卡那霉素的 1/2 MS 培养基上筛选获得 T1 植株,将筛选得到的阳性 T1 植株转到土壤中,待苗生长成熟收取 T1 代种子
转录组或荧光定量表达分析
EST(espressed sequence tag)
- 定义:表达基因所在区域的分子标签因编码DNA 序列高度保守而具有自身的特殊性质,亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有
- 技术路线:首先从样品组织中提取mRNA ,在逆转录酶的作用下用oligo ( dT) 作为引物进行RT -PCR 合成cDNA ,再选择合适的载体构建cDNA 文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST 序列的产生过程。
all right reserved @xinyi.lei
参考
https://www.jianshu.com/p/34333a6d7c4e
TBtools基因家族分析详细教程(1)会讲到修正注释文件
视频: