实验笔记:质粒的构建与表达鉴定
最近开始接触一些质粒构建和表达的分子实验,于是把本科时候做的相关实验再整理一下。
题目是:绿色荧光蛋白GFP的构建与鉴定
大肠杆菌感受态细胞的制备和质粒DNA的转化
原理:细菌处于0 ℃的CaCl2低渗溶液中,膨胀成球形,细胞膜的通透性发生变化,转化混合物中的质粒DNA形成抗DNase(脱氧核糖核酸酶)的羟基-钙磷酸复合物黏附于细胞表面,经过42℃短时间的热激处理,促进细胞吸收DNA复合物,在丰富的培养基上生长数小时后,球状细胞复原并分裂增殖,被转化的细菌中重组子基因得到表达,利用质粒上的遗传选择标记在选择培养基上可获得所需的转化子。
氯化钙法制备感受态细胞
步骤:将培养至对数生长期的E.coli DH5α(或者BL21(DE3) )在0℃经过低渗氯化钙致敏处理(预冷的 0.1mo1/L CaCl2溶液冰浴30min),就可成为高效的感受态细胞。
- 从新活化的E.coli DH5α或者 BL21(DE3)平板上挑取一单菌落,接种于5 mL LB液体培养中,37℃振荡培养过夜(12 h左右);
- 将该菌悬液以1: 100~1: 50转接于100 mL LB液体培养基中,37℃振荡扩大培养,当培养液开始出现混浊后,每隔20~30 min测一次OD600,至OD600为0.2~0.4时停止培养,并转到10 mL离心管中收集菌体;
- 培养物于冰上放置10min;
- 0~4℃,6000 g离心 3 min,弃去上清液,加入1 mL预冰冷的0.1mo1/L CaCl
2溶液,小心悬浮细胞, 将悬浮的细胞转入1.5 mL EP管中,冰浴30分钟 - 0~4℃,离心收集(菌体10000 g离心1 min或者4000 g离心10 min ),弃去上清液,加入100 µL冰冷的0.1 mol/L CaCl
2溶液,小心悬浮细胞,冰上放置片刻后,即制成了感受态细胞悬液; - 制备好的感受态细胞悬液可直接用于转化实验;也可加入占终浓度15%左右高压灭菌过的甘油,混匀后分装于1.5mL离心管中,置于-70℃条件下,可保存半年左右。
质粒DNA的转化
步骤:
每100µL感受态细胞悬液中加入1µL质粒DNA (pET-28a-GFP),轻轻摇匀,同时设置阴性对照 (质粒用等体积的无菌水代替) ;
冰上放置30 min,然后热休克(42℃水浴热激90~120 Sec,迅速冰上冷却2 min );
在超净工作台上,向上述EP管中加入0.3mL已灭菌的 LB液体培养基,摇匀后于37℃振荡培养约15~30min ,使受体菌恢复正常生长状态及表达抗性基因;
取培养液100-250µL接种于含抗菌素的LB平板培养基上,用玻璃涂布棒涂匀;
将培养皿放在37℃恒温培养箱培养30 min,待菌液完全被培养基吸收后,倒置培养皿,于37℃恒温培养箱内培养过夜(14-16 h);
观察培养皿中菌落生长情况。
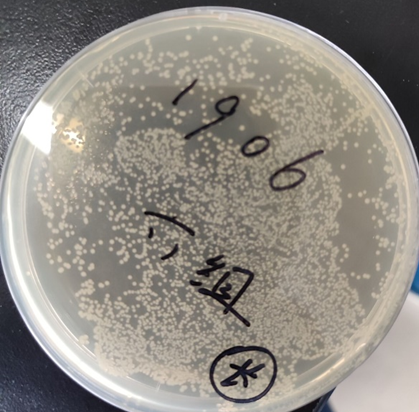
图2 Kana培养基上转化子生长情况(养了一晚上,转化还是挺多,旁边也能挑到单菌落)
碱法提取质粒DNA
原理:碱裂解法是基于大分子线性染色体DNA与小分子环形质粒DNA变性复性的差异达到分离目的;在pH 12.0~12.6的碱性环境中,线型染色体DNA和环型质粒DNA氢键会发生断裂,双链解开而变性,但质粒DNA由于其闭合环型结构,氢键仅发生部分断裂,而且其互补链不完全分离;
当将pH值调节到中性并在高盐浓度下,已分开的染色体DNA互补链不能复性,交联形成不溶的网状结构,不稳定的大分子RNA和蛋白质在SDS的作用下形成沉淀,而部分变性的闭合环状的质粒DNA中性条件下很快复性,恢复到原来的构型,呈可溶性状态保存在溶液中,离心后上清中含有质粒DNA。利用无水乙醇,吸附水分子,使DNA失水而易于聚合,可以沉淀上清液中的质粒DNA。
附:乙醇沉淀DNA需要盐离子,用金属离子屏蔽DNA上磷酸的负电荷,降低DNA分子间的斥力,才能沉淀完全。
步骤:
挑取转化筛选的带有目的质粒的大肠杆菌单菌落接种到5 mL液体培养基(LB)中,37℃震荡培养12~16 h;
将5 mL菌液转入离心管中,6000 g离心5 min收集菌体,弃上清液,在吸水纸上扣干;
离心时间不能太长,以免影响下一步的菌体悬浮
加入150mL预冷的溶液I ,于涡旋振荡器上振荡悬浮细菌细胞,尽量使细胞分散,将悬浮的细菌细胞转入1.5mL EP管;
溶液I中的葡萄糖的作用是增大溶液的粘度,减少提取过程中的机械剪切力,防止染色体DNA 的断裂;EDTA的作用是与二价离子( Ca^2+^)结合,降低DNase对DNA的降解。
加入300mL新配制的溶液II,盖紧管口,快速颠倒离心管,以混匀内容物,冰上放置3-5min;
溶液II中的NaOH与SDS可裂解细胞,使DNA变性;SDS使蛋白变性并形成交联的网状结构加入250mL
溶液III, 加盖后颠倒6-7次混匀,冰上放置2~3 min;
溶液III为低pH的醋酸钾缓冲液,中和NaOH,以便使部分变性的闭环质粒复性,而细菌染色体DNA不能正确复性
12000 g 离心5 min,取上清液500 mL移入干净EP管中,加入等体积的 Tris平衡酚/氯仿/异戊醇(25:24:1) 500 mL,缓和15 s,12000 g 离心5 min。 (平衡酚分离蛋白质及核酸,氯仿可使蛋白质变性,异戊醇防止发泡)
重复6
12000 g离心5 min,取上清液500 mL移入干净EP管中,加入等体积的氯仿/异戊醇(24:1) 500 mL,缓和15 s, 12000 g离心5 min;
将水相移入干净 EP 管中,加入2倍体积的无水乙醇,振荡混匀后,置于冰上 10min;
乙醇能够消除核酸的水化层,使带负电荷的磷酸基团暴露出来。Na离子之类的平衡离子能够与这些带电基团结合,在沉淀形成部位降低多核苷酸链之间的排斥作用。因此只有在阳离子的量足以中和暴露的磷酸残基的电荷时才会发生乙醇沉淀
12000 g离心10 min,弃上清,离心管倒置于吸水纸上扣干,得沉淀;
加500 mL 70%乙醇洗管内壁, 12000 g离心2 min,弃上清,得质粒;
A260/A280比值和琼脂糖凝胶电泳来判定质粒纯度
风干或者抽干(保存);
加TE 20μL或者灭菌水溶解质粒,加1μL RNA酶37℃ 消化30 min,放置-20度备用。
质粒DNA的限制性内切酶反应
目的:用XhoI和XbaI双酶切鉴定质粒pET-28a-GFP
将干燥并经灭菌的eppendorf管(最好0.5mL)编号,用微量移液器加样, 20 μL体系如下:
| 试剂 | 用量 |
|---|---|
| 灭菌的重蒸水 | 4.4μL |
| 质粒DNA | 10 μL |
| 相应的限制性内切酶10×缓冲液 | 4 μL |
| XhoI | 1 μL |
| XbaI | 0.6μL |
用手指轻弹管壁使溶液混匀,也可用微量离心机甩一下,使溶液集中在管底。此步操作是整个实验成败的关键,要防止错加,漏加。使用限制性内切酶时应尽量减少其离开冰的时间,以免活性降低
2)酶切
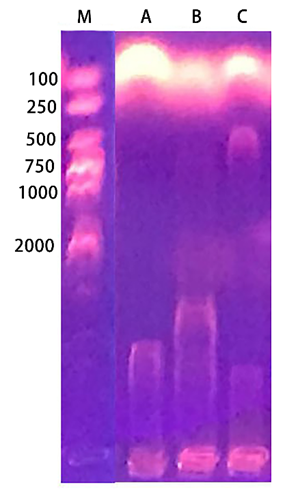
混匀反应体系后,将EP管置于适当的支持物上(如插在泡沫塑料板上),37℃水浴保温3小时(酶不同,时间不同),使酶切反应完全。在1%琼脂糖凝胶上进行电泳,染色,紫外透射仪下观察并拍照。
质粒DNA的PCR鉴定
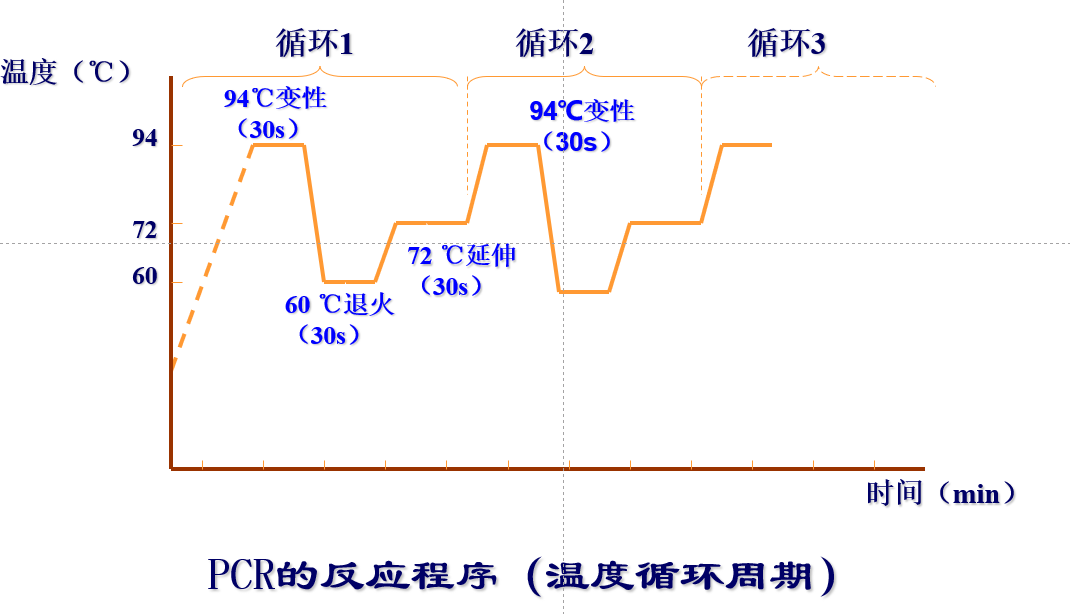
原理:聚合酶链式反应(polymerase chain reaction,PCR),是一种在体外快速扩增特定基因或DNA序列的方法,在待扩增的DNA片断两侧和与其两侧互补的两个寡核苷酸引物,经变性、退火和延伸若干个循环后,DNA扩增 2n倍
五个要素:
- 模板:单、双链DNA均可。不能混有蛋白酶、核酸酶、DNA聚合酶抑制剂、DNA结合蛋白类。一般100ng DNA模板/100mL。模板浓度过高会导致反应的非特异性增加。
- 引物浓度: 0.1-0.5 mmol/L。浓度过高易导致模板与引物错配,反应特异性下降。产生引物二聚体。
- DNA聚合酶: thermus aquaticus 0.5-2.5 U/50 mL酶量增加使反应特异性下降;酶量过少影响反应产量。
- dNTP: deoxy-ribonucleoside triphosphate(脱氧核糖核苷三磷酸)的缩写。是包括dATP、dGTP、 dTTP、dCTP等在内的统称, N是指含氮碱基,代表变量指代A、T、G、C等中的一种。dNTP浓度取决于扩增片段的长度,四种dNTP浓度应相等,浓度过高易产生错误碱基的掺入,浓度过低则降低反应产量。dNTP可与Mg2+结合,使游离的Mg2+浓度下降,影响DNA聚合酶的活性
- Mg2+:DNA聚合酶的激活剂。0.5mmol/L-2.5mmol/L反应体系。Mg2+浓度过低会使Taq酶活性丧失、PCR产量下降Mg2+过高影响反应特异性。Mg2+可与负离子结合,所以反应体系中dNTP、EDTA等的浓度影响反应中游离的Mg2+浓度。
步骤:
1)加样。按顺序将以下各成分加入PCR管中
| 成分 | 用量 |
|---|---|
| 双蒸水 | 13.4 µL |
| buffer (with Mg2+) | 2 µL |
| Mg 2+ | 0.5µL |
| pET-28a-GFP (稀释后) | 1 µL |
| 引物P1 | 0.8 µL |
| 引物P2 | 0.8µL |
| 4×dNTP | 1 µL |
| Taq 酶 | 0.5µL |
| 总体积 | 20 µL |
加好后,混匀,稍加离心并标记
2)设置PCR反应程序,将加好的PCR样品放入PCR仪中,运行PCR仪

反应结束后取10µL用1%的琼脂糖电泳进行检测(图3)
GFP的原核表达
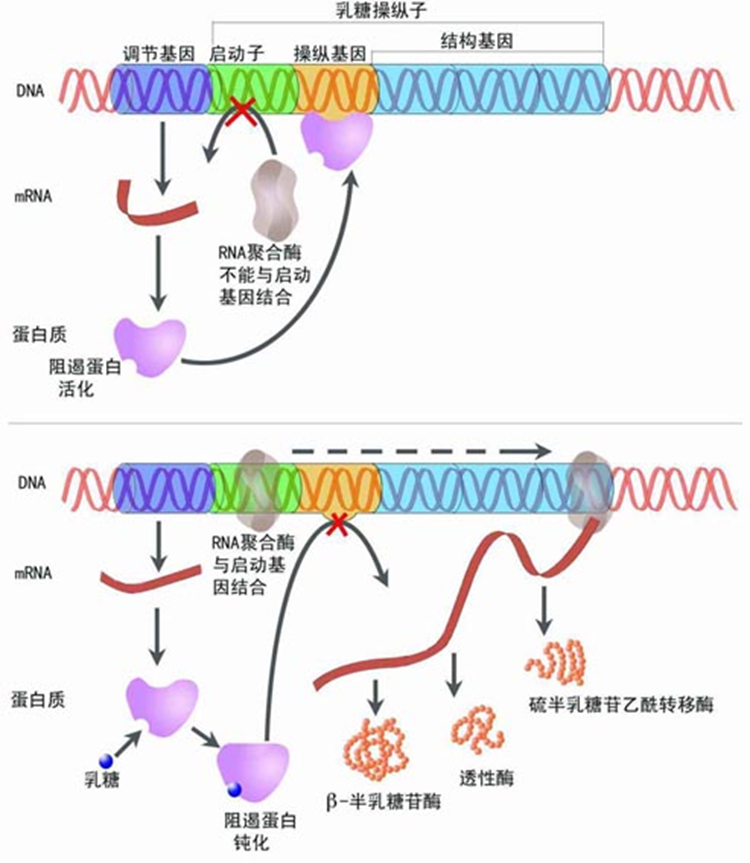
原理:将外源基因克隆在含有lac启动子的表达载体中,让其在大肠杆菌中表达。(构建);lacI产生的阻遏蛋白与lac操纵基因结合,从而不能进行外源基因的转录和表达,此时宿主菌正常生长。(转化 生长);向培养基中加入lac操纵子的诱导物IPTG,阻遏蛋白不能与操纵基因结合,则外源基因大量转录并高效表达。(表达)
BL21(DE3)菌株是在野生型BL21菌株的基础上,整合了一段来源于λ 噬菌体的DE3片段,该片段中包含了编码T7RNA聚合酶的基因,能够在IPTG或乳糖诱导下表达T7RNA聚合酶,而该聚合酶又能进一步结合T7启动子,从而启动载体上外源基因的表达。因此说,理论上只要整合了DE3片段的大肠杆菌(比如BL21(DE3)或JM109(DE3)等)都能用作pET系列载体的表达。
步骤:
- 将原核表达载体pET-28a-GFP转化到BL21(DE3)菌株中(实验方法同实验一)。
- 挑取单菌落于5mL Kana+ LB培养基培养过夜。
- 以5%的比例转接于5mL Kana+ LB培养基中,培养约3小时至OD600=0.6,即可开始诱导目的蛋白的表达。
- 加入适量的IPTG (储存浓度 200mM)至终浓度(工作浓度)为1mM,保留一管不加IPTG,作为未诱导对照,继续培养5-12小时。
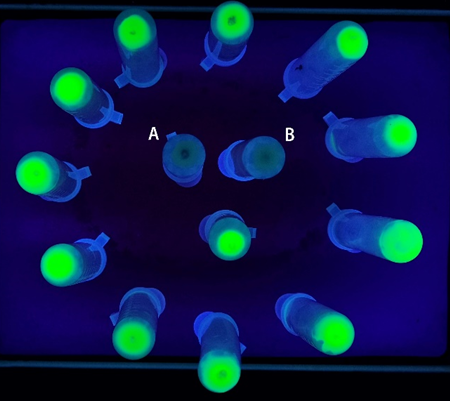
- 观察诱导表达后GFP蛋白的特征。