微生物组学分析全流程(理论)
微生物组学分析的两个大类:扩增子和宏基因组,涉及概念、流程进行区别和简单介绍。
引言
DNA测序技术
| 测序技术 | 方法 | 优点 | 缺点 |
|---|---|---|---|
| 第一代测序技术 | Sanger测序技术 | 准确性高 | |
| 第二代测序技术 | 高通量测序技术(不同平台) | PCR引入的系统偏差和随机性 | |
| 第三代测序技术 | 纳米孔等 | 1. 没有PCR带来的不确定性和偏差 2. 速度和长度得到提升 | 较高错误率 |
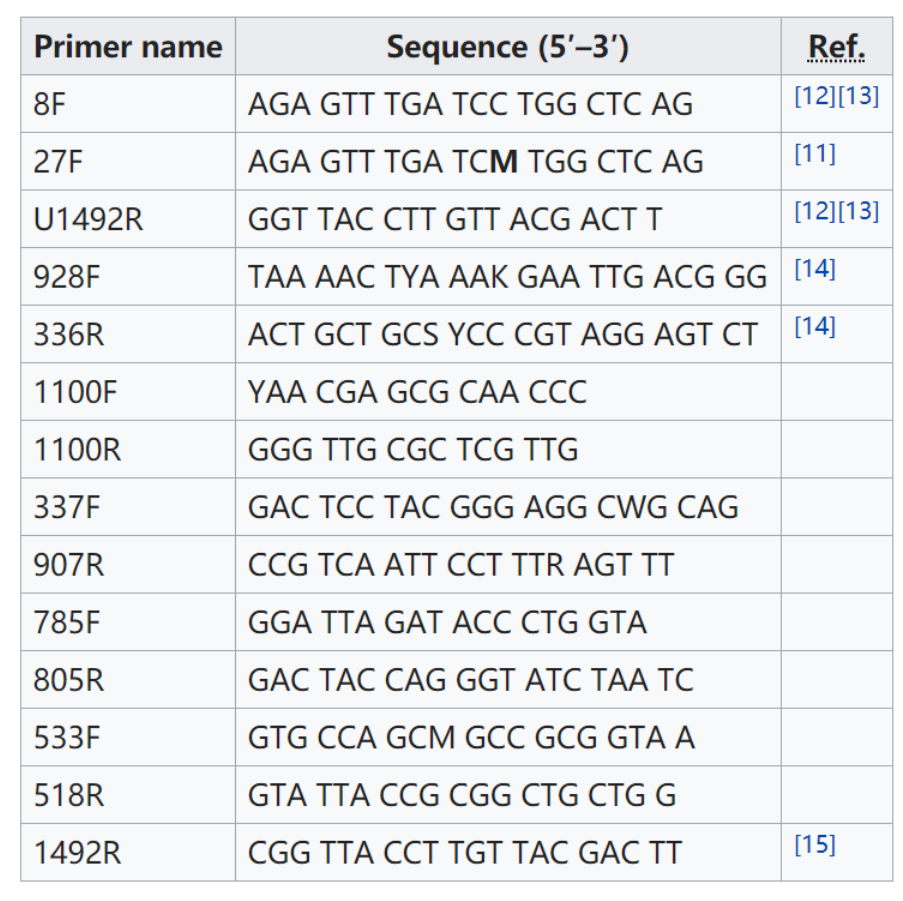
常用引物
测序数据存储格式
无论哪种测序技术或机器平台,测序数据一般都会以特定格式的文本文件进行存储。以 FASTQ 格式为例(如图 1.2 所示),其中每一条 DNA 序列 包含 4 行信息:第一行以字符“@”开始是序列的唯一标识;第二行是 DNA 序列的碱基信息;第三行是字符“+”,可用于添加对序列的额外描述;第 四行中每个字符是对应 DNA 碱基的测序质量评估分数。
碱基的测序质量评估分数:
- 质量值体系分为 Phred33 和 Phred 64两种
- 测序过程是一个化学反应过程,随着试剂或者实验环境等因素的变化,可能导致机器对碱基识别的错 误。基于 DNA 测序的研究中普遍会存在测序错误,并不仅仅局限于 16S rRNA 的研究。因此,常见的测序平台会对每个碱基的测序质量打分并且将质量分 数与 DNA 序列信息一起输出。Illumina 平台下就是使用 Phred 质量分数
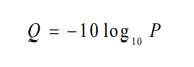
- 其中 P 表示该碱基识别错误的概率。因此如果一个碱基的 Phred 分数为 30 则表示该碱基是测序错误的概率为 0.001。在 FASTQ 格式中,Phred 分数往 往被编码成 ASCII 码对应字符。实际数据处理中,通过 FastQC[57]先对包括 Phred 分数在内的多种质量指标做评估,生成 HTML 格 式的质量报告。据此,再通过 FASTX-Toolkit对测序数据做质量控制,包括(1)剔除含有未识别碱基(FASTQ 文件中用字符“N”表示)的序列; (2)根据序列的最低、平均或者类似准则来剔除低质量序列。
16S rRNA 测序和宏基因组测序
扩增子测序是指利用合适的通用引物扩增环境中微生物的16S rDNA/18S rDNA /ITS高变区或功能基因,通过高通量测序技术检测PCR产物的序列变异和丰度信息,分析该环境下的微生物群落的多样性和分布规律,以揭示环境样品中众多的微生物种类及它们之间的相对丰度和进化关系。
宏基因组测序所得信息包括:微生物整个基因组而不仅仅是特定基因,理论上能够提供更加精准的物种分类
16S测序:测它的16S对应的DNA序列,尽管这段区域在不同物种间差异度较高,但不能保证将所有的细菌种类完全鉴定区分
宏基因组测序:测全部DNA信息,但是贵
所以一般,先用16S缩小范围,再宏基因组
16S rDNA测序
【1】Woese CR. Bacterial evolution. Microbiological reviews.1987, 51:221 -271
指出核糖体核糖核酸(ribosomal RNA)作为研究生物进化关系“分子 钟”的良好适用性
核糖体的主要成分是蛋白质(40%)和 RNA(60%),按沉降系数可以 分为 70S(原核)和 80S(真核) 两类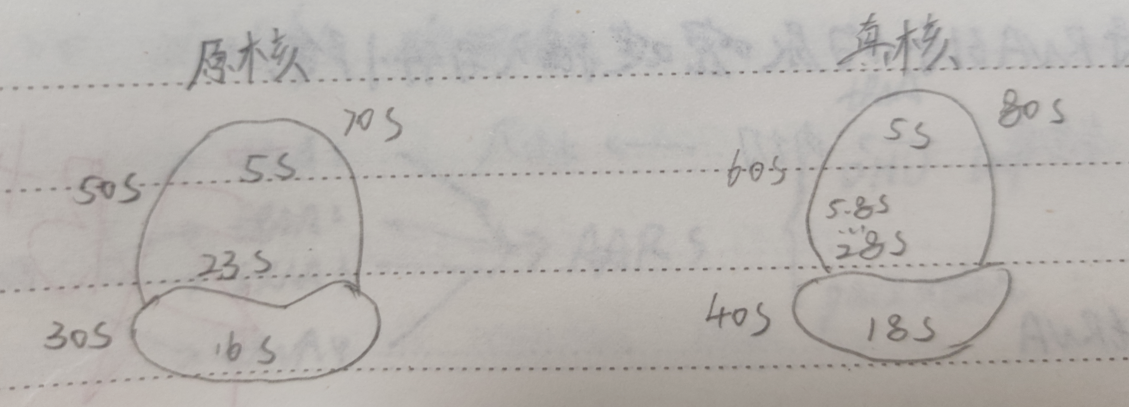
测序区域选择
原核(细菌)测序一般选择16S测序,16s rRNA基因有10个保守区和9个高变区(v1-v9:长度分布范围约30~100bp),可以测全长,也可以只测碱基变化率较高的部分区域如V3-V4区
选哪个区域与具体研究需求和测序平台的读长有关,比如 Illumina Hiseq2000 平台只有 101-151 的长度,因此常常选用 V6 区域
真菌一般选择ITS,也会ITS和18S联合使用,提高准确度,区域一般为ITS1-ITS2
藻类选择18S
真核rRNA加工如下图
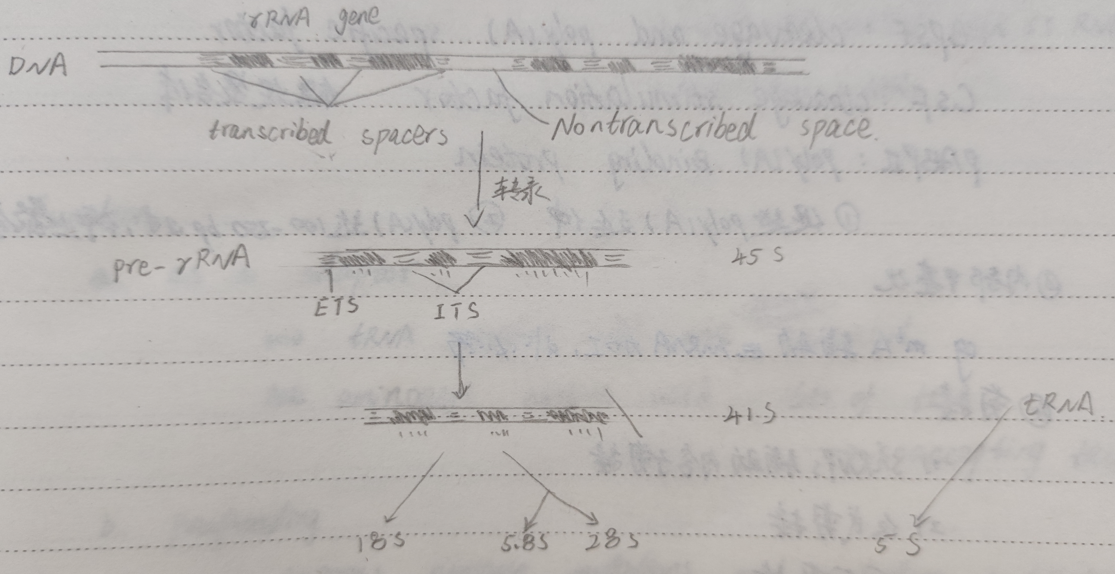
由于 RNA 稳定性差,在实验 操作分离、纯化等过程中容易因降解而丢失信息,研究者倾向于对 16S rRNA基因(DNA)进行测序。
但是,16S rRNA 的分类精度是有限的。通常情况下,生物学分类系统 包括界(kingdom)、门(phylum)、纲(class)、目(order)、科(family)、 属(genus)和种(species)等七个分类水平,且分类细致程度逐渐提高②。 一般情况下,基于 16S rRNA 基因序列能够精确分类到 genus 水平,对于 species 水平的分类却不够可信
物种多样性
无论以何种方法进行微生物群落研究,多数工作中研究者主要关注环境 中微生物的生物多样性(biodiversity),包括物种多样性(species diversity) 和遗传多样性(genetic diversity)两方面。
物种多样性包括
Alpha 多样性:种内差异
Beta 多样性:种间差异
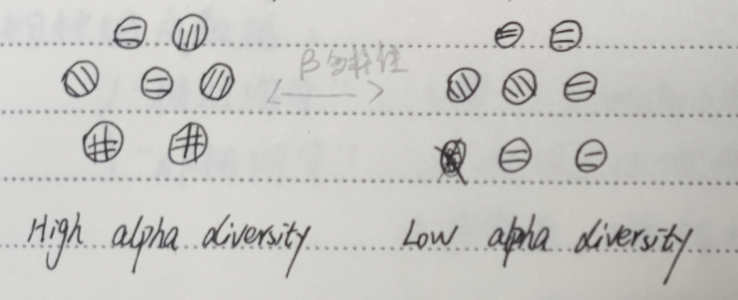
Alpha 多样性
多样性指数
丰富度:Chao 1 \ACE \ Observed species
多样性:Shannon\Simpson
上面都是基于OTU的,还有基于进化树的指数:Faith’s PD
测序
多样性指数的计算只是在当前测序深度(样本的测序所得序列总数)下所得, 测序数据中可能并没有包含全部的物种信息。因此,真实的 Alpha 多样性需 要通过一定模型进行估计。

Beta 多样性
由于测序的随机性,不同样本的测序深度 不同会导致 Beta 多样性评估有系统性偏差。因此,在 Beta 多样性指数计算 之前往往需要对样本的测序深度进行归一化。
微生物组学的发展
项目
2007年,美国和欧盟,人类微生物组计划,HMP(Human Microbiome Project)
2010年,欧盟,人类肠道宏基因组计划,Meta-HIT(Metagenomics of the Human Intestinal Tract)
2014年,综合人体微生物组计划
2016年,美国,国家微生物组计划,MNI(Nationl Microbiome Initiative)
2016年12月1日,中国微生物研究计划
6个领域:人体、环境、农作物、家养动物肠道、工业及海洋
两个方向:微生物组研究方法及应用技术平台和微生物组数据储存及功能挖掘
软件
Qiime2 :以 Python 脚本将已有软件的输入输出 文件格式“对接”,从而直接集成以实现其定义的流程
基于 16S rRNA 基因测序数据的处理
主要包括测序质量控制、双端序列连接、混合样本分离、OTU 划分和丰度信息计算以及分类信息注释。测序质量控制主要是根据 Illumina 测序平台给出的碱基质量分数来剔除错误率较高的序列;双端序列通过重叠 的区域进行连接达到延长序列的目的;混合样本分离是依靠嵌入在序列中的 人工编码序列;OTU 划分是将 DNA 序列进行聚类得到分类学单元,作为后 续统计分析的特征单元;OTU 的丰度信息由序列数量计算得到,分类信息 则是通过公共数据库的比对来确定。
理论
实验部分
总DNA提取
核酸裂解提取总微生物基因组,提取的 DNA 采用分光光度计,测量在 230-260-280nm 的吸光度,检测 DNA 纯度和浓度, 所有提取的总DNA 样品在测序之前,均密封在-70℃的冰箱备用。
- DNA浓度:≥10ng/ul
- DNA纯度:A260/A280 = 1.8 – 2.0左右
- DNA总量:≥300ng
- DNA完整性:要有明显的基因组主带
文库构建
根据保守区设计得到引物,在引物末端加上测序接头,进行PCR扩增并对其产物进行纯化、定量和均一化形成测序文库
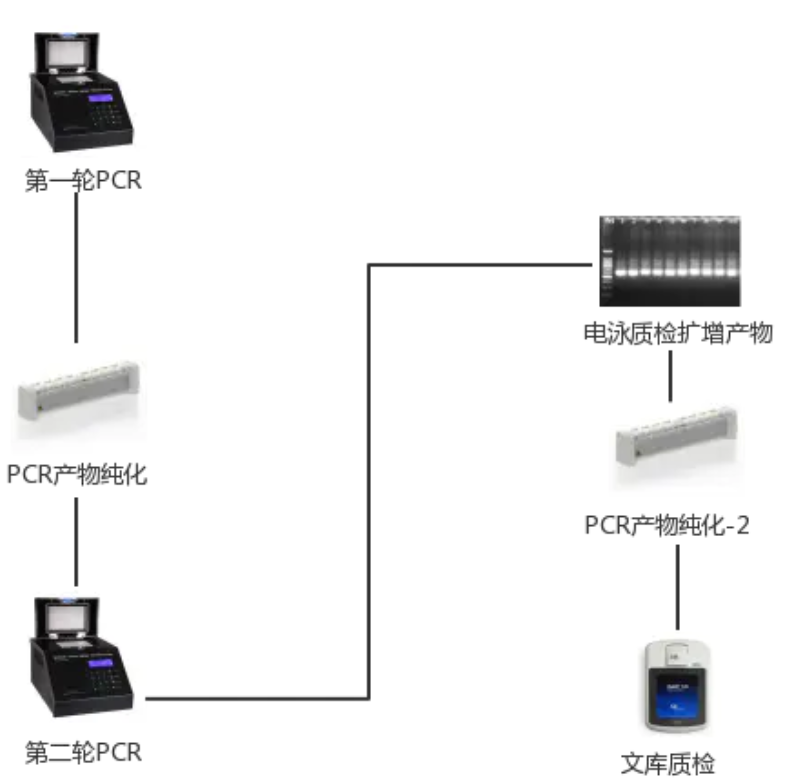



质检
建好的文库先进行文库质检,质检合格的文库用Illumina HiSeq 2500进行测序。
测序得到raw data
Illumina二代测序的具体过程可以通过官方视频了解,十分详细。大概是:
- 采集样品(如粪便,皮肤等), 提取微生物DNA
- 扩增DNA:通过桥式扩增,得到大量扩增的DNA片段
- 测序:将带荧光标记的叠氮基团结合到待测链上,得到各碱基对应的特定荧光,以此得到测序结果。
- output: 测序得到n个reads,被output成fastq文件,根据primer和barcode来整理这些reads,得到各个样本的R1和R2,再进行后续分析。
高通量测序(如Illumina HiSeq等测序平台)得到的原始图像数据文件,经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(Reads)的序列信息以及其对应的测序质量信息。
一、数据预处理
测序的结果会保存为通用的 FASTQ 格式,以下处理都基于这个测序数据
送公司测序返还的数据一般都是拆分过并去除了引物的,可以自己再做一下质检,后续使用dada2分析时也会有碱基质量分布图,所以这步可以不做,自己质检(fastqc)的信息会比较全面。
1.混合样本分离合并
由于测序实验设计中为了降低成本,往往将多个样本混合后再一起测序,其中为了区别不同样本会在 DNA 分子中用实验方法嵌入一段人工合成的编码序列(barcode)。混合样本的分离就是靠识别该端 barcode 序列,将一个 FASTQ 文件中的序列分别存储成为多个 FASTQ 文件,分别保存属于不同样本的序列。
列举工作中常见的两种双端测序得到的raw seqence data:
1)未分样本,按批次分文件的下机序列数据:
批次a_R1.fastq.gz, 批次a_R2.fastq.gz
批次b_R1.fastq.gz, 批次b_R2.fastq.gz …
这样的文件会附一个mapping file, 提供了各个样本的barcode,ForwardPrimer及ReversePrimer,如下所示。有的只有一边barcode,有的有双barcode,下表为双barcode的例子:
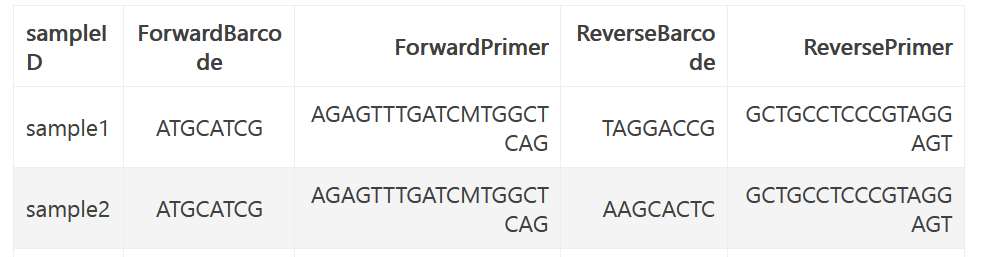
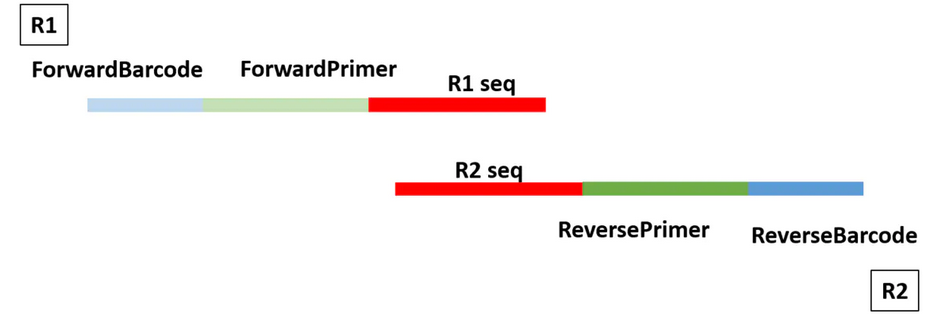
示意图

fastq格式 : 比方说这是a批次的R1和R2
2)按样本分好各文件的序列数据:
sample1_R1.fastq.gz, sample1_R2.fastq.gz
sample2_R1.fastq.gz, sample2_R2.fastq.gz …
这种data不是很raw,是将1中的下机数据按照mapping file中样本的信息分好,则得到这样的文件。由于分好样本了,基本不需要mapping file。但如果没有去除adapter和primer,你需要找测序的人要adapter和primer,并且grep一下看看到底有没有。
其fastq格式和上面的差不多。在不同的流程和软件处理的过程中,其header会根据分析需求做出改变。
2.质检
fastqc
3.切除引物
4.质控
5.双端序列拼接
当待测 DNA 分子长度小于测序平台读长 2 倍时,双端序列会有重叠部分;通过重叠部分将测序得到的双端序列数据拼接 (Merge)成一条序列Tags。
会有一些算法用于确定重叠区域位置,主要根据碱基匹配率确定
二、可操作分类单元(OTU)划分
通过序列 比对将满足相似度阈值的 16S rRNA 基因序列聚类而得到的序列集合。
意义
- 减少后续分析的计算量
- OTU在聚类过程中会去除一些测序错误的序列,提高分析的准确性。
方法
层次聚类法
计算序列 k-mer差异程度,过滤掉不必要的比对
k-mer 差异较大的序列不太可能有较高的序列相 似度
但该类方法的计算量还是太大,不适用于大 规模数据
基于序列两两比对得到距离矩阵
指定阈 值进行层次聚类
基于统计模型的方法
认为测序是采样过程,OTU 中序列之间的相似度 服从某一概率分布,而每一条 DNA 序列都是对该分布的采样结果。主要思想就是将 聚类问题转换为概率分布的参数估计问题,但是因为参数估计的计算复杂度 使得该类方法同样不适用于大规模数据。
启发式聚类
通过特定准则选 定一些序列作为初始种子,将待聚类的序列按指定标准进行排序。聚类过程 中,待聚类序列仅与种子进行比较来决定该序列是否属于某一聚类或者成为 新种子,因此大大减少了序列比对的次数。启发式方法的贪婪策略使聚类结 果不是最优,但能够适用于大规模数据集。根据是否使用参考序列,启发式聚类方法可分为两类。
基于参考序列(reference-based)
是将序列比对到公共数据库,比对结果一致的所有序列集合成为一个 OTU,同时比对到的参考序列的分类信息即作为该 OTU 的分类注释信息。对于不能比对到数据库的序列不再进一步处理,因 此这种 OTU 划分方法适用于未知微生物较少的环境。
不基于参考序列(de novo)
能够对有所有的序列进行 OTU 划分,聚类初始的种子一般选 择长度较长或出现次数较多的序列,之后的聚类过程与上文描述一致。de novo 聚类方法贪婪的策略不保证最优,一些研究者为了提高 OTU 划分的准 确度在聚类过程中引入了合并(merge)和分裂(split)操作。由于单一序列与一 个类的相似度≥97%并不一定保证该序列与类中的每一条序列距离都≥97%, 因此随着聚类的进行,一个类中序列之间的差异可能逐渐增大。当差异超过 3%时,这个聚类就需要被分裂成两个聚类才合理;同理,有时也需要对一 个 OTU 进行合并。
为了充分利用测序数据,会将两者结合起来使用。2014年最新发布的Qiime中,提出了称为subsampled open-reference clustering的OTU划分策略
(1)首先使用较低的相似度(如60%)进行第一次reference-based的OTU 划分,比对不上的序列即直接舍弃,理由是这些序列可能只是测序错误而不 是真正有生物意义的16S rRNA基因片段;
(2)然后,使用97%的相似度阈值再次进行reference-based的OTU划分;
(3)对于(2)中没有比对到数据库的序列,首先随机采样0.001%的序 列进行一次de novo聚类,并从所得OTU中挑选出相应的代表序列;
(4)以所得代表序列作为参考序列,对(2)中没有比对到数据库的所 有序列进行de novo聚类;
(5)最终将(2)、(4)中所得OTU合并即成为最终的OTU划分结果。
总结:挑选非重复序列→与16S数据库比对→去除嵌合体序列→序列之间距离计算→97%相似OTU聚类
嵌合体序列是RCR扩增时,两条不同的序列产生杂交、扩增的序列。
三、物种分类及丰度信息注释
对于物种注释的算法,目前主流的有RDP classifier ,SINTAX , Mycofier,MOTHUR。
- RDP classifier基于贝叶斯算法(naïve Bayes algorithm),并基于k-mer (k=8)提取序列特征。
- SINTAX使用非贝叶斯算法(non-Bayesian classifier),并基于k-mer (k=8)提取序列特征。
- MOTHUR使用k-nearest neighbor (kNN),并基于k-mer (k=8)提取序列特征。
- Mycofier使用贝叶斯算法(naïve Bayes algorithm),并基于k-mer (k=5)提取序列特征。
Qiime 策略:将 OTU 中的所有序列用 UCLUST 算法比对到 Greengenes 数据库,如果超过 50%的序列都能一致地比对到同一分类单元,则将该分类信息作为 OTU 的注释。
RDP\SILVA\Greengenes是三个主要的 16S rRNA 基因公共数据库
- 16S、18S 和功能基因默认采用RDP classifier贝叶斯算法,ITS和AMF默认采用Blast方法,置信度阈值为0.5。
- 16S(细菌、古菌)和18S(真核)默认用silva,ITS(真菌)默认用unite,功能基因默认用fungene(RDP整理来源于GeneBank的功能基因数据库),AMF默认用MaarjAM数据库,得到物种注释信息表otu_taxa。
虽然可以只 将 OTU 的代表序列进行比对,但在 OTU 内序列较多时,一条甚至多条序列 都不能很好代表 OTU
实操
check list:拿到raw sequence data后,开始上游分析前
手头的数据测的是16s的哪个区域?v1-v2?v3-v4?v4?,check对应的primer. 将For和Rev的primer在R1和R2的序列中都grep一下。checkR1的file中是否存在ReversePrimer,R2的file中是否存在ForwardPrimer。 如果存在这种“互换”的情况,需要把它们换回来。并且记得将header中R1,R2的信息更新。
raw data分好样本了吗?没有的话需要根据barcode和primer来分样本,跟测序合作者要mapping file mapping file获取之后,随机抽取一些barcode和primer检查,防止出现1中的问题。以及要检查primer前的的序列是否真的是barcode。barcode前面是否还有其它前缀序列。
分好样本的为XXR1.fastq, XXR2.fastq这样的格式,不论是你自己分的还是raw data给到你就是这个,都要check primer和barcode是否还在上面,决定后面是否需要cutadapt。自己cutadapt之后也要check一下,以防cut错了。
在R1和R2中随机抽取几条blast看一下方向,是否与R1(+), R2(-)一致。也是防止R1和R2发生“互调”的问题。
数据预处理
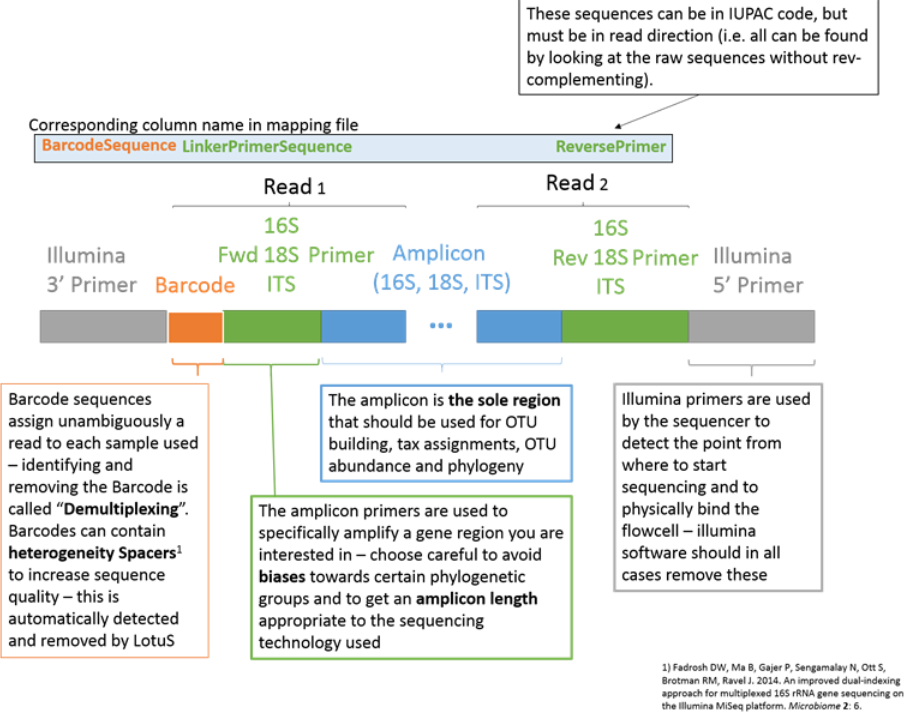
需要先了解barcode, primer的概念,可以通过下图快速了解一下:barcode是每个样本对应的一段与样本一一对应的短序列,像超市货物条形码一样区别每个样本。Fwd和Rev primer(引物)是用于16S扩增与建库的引物序列,在测序技术中起到定位的作用。有些下机数据会已经将样本按照barcode分好,每个样本测得的序列片段都放在一个与样本对应的文件里。
基于宏基因组测序数据的处理
生物实验提取得到宏基因组 DNA 分子后,先“打 断”成指定长度的片段再进行测序。由于本文主要关注微生物群落的物种多 样性分析,因此数据处理仅包括测序质量控制、“污染”和宿主序列排除、 物种丰度和分类信息获取等三部分。宏基因组测序的质量控制与 16S rRNA 基因测序略有不同,除了剔除整条序列还需要对序列中部分低质量碱基裁剪 丢弃。所谓的“污染”序列是指生物实验过程中人为因素引入的不属于目标 微生物群落的 DNA 序列;而宿主序列是由于采样过程不可避免地包含了宿 主细胞,从而在宏基因组提取中夹杂了宿主 DNA 信息。通过宏基因组测序 数据得到物种丰度和分类信息则有多种方法,本文中采用直接将序列比对到 事先建立的标识基因数据库。相比与所有已知微生物的整个基因组进行比对, 该方法更加有效率,而且分类信息注释结果的一致性较好
实验部分
DNA提取
核酸裂解提取总微生物基因组,提取的 DNA 采用分光光度计,测量在 230-260-280nm 的吸光度,检测 DNA 纯度和浓度, 所有提取的总DNA 样品在测序之前,均密封在-70℃的冰箱备用。
将DNA分子打断成多个片段。
打断的原因是测序读长不够
测序
一、数据预处理
1.质量控制
(1)剔除含有未识别碱基(FASTQ 文件中用字符“N”表示)的序列;
(2)根据序列的最低、平均或者类似准则来剔除低质量序列。
(3)序列剪裁:从序列中截取出满足指定质量分数阈值的最长连续片段。
16S rRNA 基因测序数据处 理中,由于在 OTU 划分时一般采用固定阈值(如 1.2.2 节中说明),而且被 测序高变区的长度往往在一两百个碱基左右,序列中增减几个碱基会对相似 度有较大影响。因此,在 16S rRNA 基因序列的质控上一般不会对序列进行 裁剪。宏基因组测序数据是基因组的序列,后续处理中往往需要进行拼接, 低质量的序列末端会导致很多拼接错误,因此往往需要进行裁剪。
2.“污染”序列剔除
16S rRNA 基因测序的前期实验是通过“探针”序列锚定以定向获取 DNA 序列,而宏基因组在“打断”宏基因组 DNA 分子后即可直接测序,因此数 据中难免会包含不属于微生物群落的序列,如人体环境宏基因组中的人的 DNA。本文中采用 SOAPAligner[70]比对工具将测序数据比对到人类基因组数据库,能够比对上的序列就舍弃不用。而剩下的不能比对上的序列就认为是 属于微生物的序列,进入后续分析流程。同类研究中去除“污染”序列的方 法基本大多基于参考序列比对,差异仅是使用的比对软件和数据库不同,如 常见的短序列比对软件还有 BWA[71]、Bowtie2
3.不需要双端序列拼接
宏基因组测序也是使用双端测序策略,然而不同的是,这里的双端序列 目的主要是为了跨越更大的基因组区域,因此往往一对序列不对有重叠部分, 所以预处理中不需要有双端序列拼接的步骤。
二、物种分类及丰度信息注释
考虑到拼接所得长序列本质 是来源于原始测序的短序列,因此理论上用短序列直接进行比对应该与比对 拼接后的长序列有一致的结果。
在早期的宏基因组测序研究中,经过预处理步骤之后往往需要经历短序 列拼接的过程。因为只有通过拼接之后,才能够得到更完整的基因序列。
软件 Metaphlan分类方法:
建立标识基因数据库
以 NCBI 数据库中记录的 2887 个微生物基因组和对应的物种进化树为输入,通过基 因组之间的序列比对,从种(species)的分类水平开始找对于每个分类单元唯一特异的基因,一直到门(phylum)级别分类水平
标识基因,反映了基因组中不仅仅只有 16S rRNA 基因可以用来物种分类
比对并统计
将测序数据比对到标识基因数据库 并且计算在不同分类水平下各个分类单元的丰度