rna-seq上下游分析实战
从零开始系列。网上的流程都太散乱了,无法连接起来,本文一套数据跑完流程,包括转录组基本介绍、上游数据处理和下游功能分析
关于生物信息学,我比较推荐的一些在线书:
在线操作平台:
- 国家基因组科学数据中心
- Galaxy 比较全面,有许多小工具,但有时你可能得需要vpn
在开始之前,你也许可以先看看:
- 生信宝典分享的一篇Nature综述:正文 | 笔记
- 转录组学习路径:RNA-seq分析学习路径图
- 转录组分析流程:
- 文字 : 张一柯 转录组分析流程
- 代码 : 生信技能树 转录组分析上下游
- 统计学知识:RNA-seq中的那些统计学问题
前言
这部分主要补充一些背景知识。分析流程可以从2. 转录组分析开始看
多组学分析概况
高通量测序技术的发展,使得多种组学分析得以迅速发展,常见的组学研究有:基因组学、转录组学、蛋白组学、代谢组学。此外还有表观基因组、脂质体和微生物组等,这里我们不深入分析。下面我们主要讲一下四大基本组学。
基因组学
顾名思义,基于DNA测序,研究对象包括基因组的结构、功能、进化、定位、编辑等
转录组学
转录组是特定发育阶段或生理条件下细胞内的完整转录信息的集合,代表了基因表达的中间状态。基因组在所有的细胞中几乎都是完整的,与之不同,转录组具有较高的细胞和组织特异性。有利于研究胁迫等。
对于转录组,可以从两个方面来定义:(1)广义转录组:指生物体细胞或组织在特定状态下所转录出 来的所有 RNA 的总和,包括能够编码蛋白质的 RNA (即 mRNA)和不能编码蛋白质的 RNA (ncRNA, 如 rRNA, tRNA, mieroRNA 等);(2)狭义转录组:通常是指所有 mRNA 的总和。一般文章中无特殊说明都是指mRNA。
研究方法:
- 基于实验:Northern blotting; RT-PCR
- 基于测序:
- 最开始用的EST:mRNA反转录形成cDNA,通常不能形成全长,其片段就称为EST
- 然后有了基于杂交的基因芯片:需要参考转录组信息,利用碱基互补配对,将荧光标记的cDNA制成微阵列探针。
- 现在用的就是NGS啦,也就是我们称的RNA-seq
蛋白组学
生物体内执行功能的最终载体,在转录时,不同的转录起始和mRNA剪切使同一基因产生了不同的转录产物。同样由于不同的翻译起始,一个mRNA可以翻译成不同的蛋白质。蛋白质还可通过翻译后的修饰,如磷酸化( phosphorylation)、甲基化( methylation)、泛素化( ubiquitylation) 或蛋白质降解等过程,改变蛋白质的活性、细胞定位和蛋白质的稳定性。
蛋白质组研究是指系统研究在某一特定时间、特定条件下,某一种特定组织中的所有蛋白质。对蛋白质的研究也不只是局限于蛋白质的氨基酸序列,而是包括了蛋白质的表达量,蛋白质活性,被修饰的状况以及和其他蛋白质或分子的相互作用情况、亚细胞定位和三维结构。
研究方法:
- 定性与纯化:凝胶技术和质谱技术
- 定量:在获得蛋白标志物或者肽标志物后,可通过液相色谱-质谱的选择反应监测(SRM)
代谢组学
以生命体的代谢物为研究对象,主要研究分子量1000以下的小分子。根据研究对象不同,代谢组学可以分为研究已知化合物的靶向代谢组学和分析未知化合物非靶向代谢组学。
常见的代谢物:主要有极性化合物(例如有机酸、氨基酸、糖、胺)、脂类、类萜和固醇。
研究方法:
- 核磁共振技术(NMR):具有非破坏性的优点,可以对研究对象内部化学变化和生化反应进行跟踪
- 质谱
实验设计
基本原则
- 生物学重复排除随机误差
- 样本提取时控制干扰变量。由于基因表达的时空特异性,在样本提取的时候要注意提取时间和组织细胞的控制,以及提取后样本的妥善保存(及时冷冻等)
- 测序深度根据实验具体要求而定。如果需要鉴定新转录本或没有参考基因组,那么就适当增加测序深度;对于small RNA同样要适当提高测序深度,一般需要30M以上的有效匹配reads。
- 文库构建分为链特异性文库和非链特异性文库。链特异性文库可以区分转录本的方向信息以区分转录本来源,避免互补链干扰
- 测序策略包括单端和双端测序。单端测序只在cDNA一侧末端加上接头,引物序列连接到另一端,扩增并测序。而双端序列会在cDNA片段两端都加上接头和引物,进行两轮合成测序。

转录组文库制备
Total RNA
一般生物体中的的 RNA中,rRNA占绝大多数,含量超过 90%,而mRNA的含量在 1-2% 左右,转录组测序文库是以样本的Total RNA为基础,从中提取mRNA构建测序文库,因此文库构建包括mRNA富集和碎片化、mRNA反转录、接头添加和PCR富集等过程
RNA是单链,DNA是双链,能够进行碱基互补配对,从而测序

mRNA富集
在生物的Total RNA中,mRNA所占比例很低,绝大部份时核糖体RNA (rRNA),因此如果直接使用Total RNA进行测序,得到的结果必然是不准确的,因此需要先将Total RNA中的mRNA分类纯化出来。需要注意的是mRNA极易被RNA酶降解,故在提取时要注意防止样品引入RNA酶污染。
转录组测序的文库根据待测样品的物种分为真核转录组和原核转录组。由于真核生物和原核生物mRNA结构不同,因此在mRNA富集时所采用的方法也并不相同。
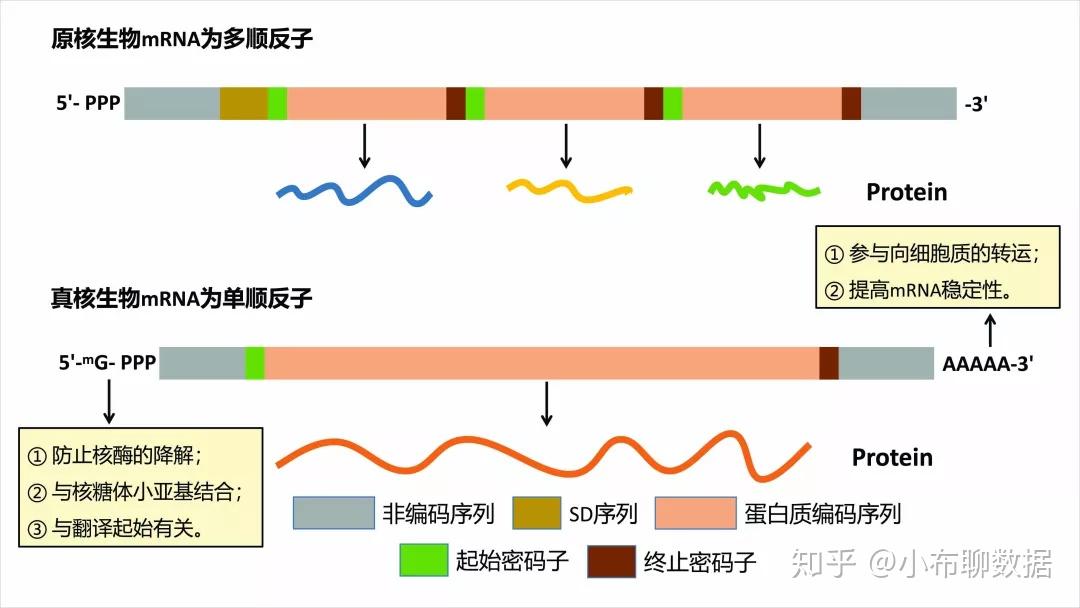
真核生物mRNA中含有polyA尾结构,因此可以使用oligoT与其特异性的匹配,从而在Total RNA中特异性的富集mRNA。
然而,原核生物并不像真核生物mRNA具有polyA的结构,因此,无法直接利用oligoT将mRNA纯化出来,目前,提高原核生物中mRNA的量,最主要的方式是去除Total RNA中rRNA。
链特异性文库
在于合成第二链cDNA时,用dUTP代替dTTP,此时第二链cDNA上布满了含dUTP的位点。这样就可以区分cDNA中的正义链和反义链,保留mRNA单链的特性。在接头连接后用一种特定的酶,其可在尿嘧啶位置产生一个单核苷酸缺口,从而消化掉第二链cDNA,只保留第一链cDNA,随后进行PCR扩增纯化,得到只含第一链cDNA信息的文库。
总结
- 取样,设置合适重复
- PolyA(或rRNA移除)富集分离纯化RNA
- 随机打碎成短片段并反转录为cDNA,长度筛选后添加接头构建文库
- PCR扩增并测序
测序技术
测序基本流程
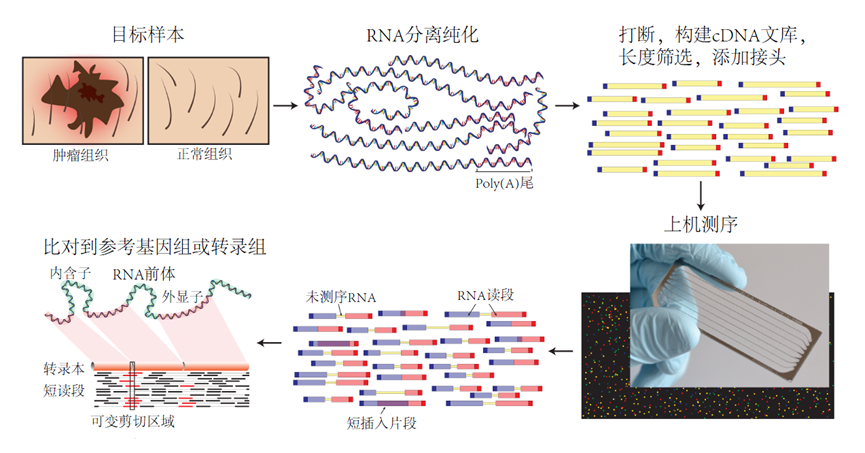
第一代测序技术
双脱氧末端终止法(Sanger法)
原理:ddNTP 的2′和3′没有羟基。在DNA合成的过程中,需要3′和5′端进行羧化反应形成磷酸二酯键,从而形成 DNA 链,由于ddNTP的插入,没有3′羟基的存在,阻碍 DNA 的进一步生成。从而形成一系列长度不等的核酸片段(长度相邻者仅差一个核苷酸)。
具体操作:将4种不同碱基(A、T、C、G)用4 种不同的放射性同位素进行标记,在一定比例4种DNA 聚合酶的作用下进行反应,之后进行凝胶电泳,再通过放射自显影技术判断合成片段的长短以及碱基类型,依次可以得出DNA片段碱基序列。
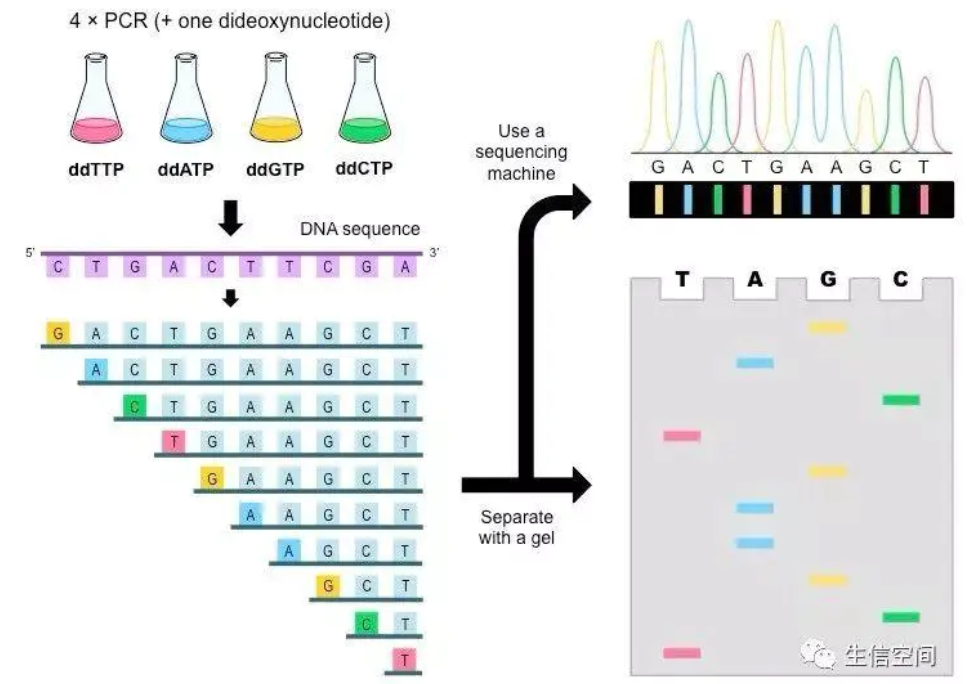
更多介绍可以看这篇:DNA测序——双脱氧末端终止法(Sanger法)
第二代测序技术
高通量测序(High-throughput sequencing)
是基于PCR和基因芯片发展而来的DNA测序技术。我们都知道一代测序为合成终止测序,而二代测序开创性的引入了可逆终止末端,从而实现边合成边测序(Sequencing by Synthesis)。二代测序在DNA复制过程中通过捕捉新添加的碱基所携带的特殊标记(一般为荧光分子标记)来确定DNA的序列。核心技术是DNA合成的可逆性末端循环,即3’-OH可逆性的修饰和去修饰。将dNTP的3’-OH以叠氮集团RTG(Reversible Terminating Group,可逆末端基团)进行修饰;将4种碱基分别与不同的荧光分子连接;DNA合成时,RTG能起到类似于ddNTP的作用终止反应;每次合成反应终止并读取信号之后,洗脱RTG和荧光分子,进行下一轮循环。
平台:主要包括Roche的454 FLX、Illumina的Miseq/Hiseq等。
缺点:在二代测序中,单个DNA分子必须扩增成由相同DNA组成的基因簇,然后进行同步复制,来增强荧光信号强度从而读出DNA序列;而随着读长增长,基因簇复制的协同性降低,导致碱基测序质量下降,这严格限制了二代测序的读长(不超过500bp),因此,二代测序具有通量高、读长短的特点。二代测序适合扩增子测序(例如16S、18S、ITS的可变区),而基因组、宏基因组DNA则需要使用鸟枪法(Shotgun method)打断成小片段,测序完毕后再使用生物信息学方法进行拼接。
文字: 第二代测序原理的详细解析
第三代测序技术
单分子测序技术
在测序过程中不需要涉及PCR扩增,实现了对每一条DNA分子的单独测序。三代测序技术具有超长读长,还拥有不需要模板扩增、运行时间较短、直接检测表观修饰位点、较高的随机测序错误等特点。它弥补了第二代测序读长短、受GC含量影响大等局限性,已在小型基因组从头测序和组装中有较多应用。
目前比较有代表性的三代测序平台公司有三家,分别是Pacific Biosciences(PacBio)公司的单分子实时测序技术、Oxford Nanopore公司的单分子纳米孔测序技术、和Helicos公司的真正单分子测序技术tSMSTM。
纳米孔测序
第一、二、三代测序技术都是基于边合成边测序的原理,因此Nanopore技术被一些人称为第四代测序技术;
采用电泳技术,借助电泳驱动单个分子逐一通过纳米孔来实现测序的。由于纳米孔的直径非常细小,仅允许单个核酸聚合物通过,而ATCG单个碱基的带电性质不一样,通过电信号的差异就能检测出通过的碱基别,从而实现测序。
测序深度与覆盖度
概念:点击此处了解
要求:有参,不进行可变剪切或新转录本的分析和检测,则测序深度可以稍微低一些
总结
四大测序技术的优缺点:
Sanger法测序读长长、准确度高,但是通量不高;
Illumina测序读长短、通量高、准确度高,在进行基因组组装或者结构变异分析的时候没有优势,可用作三四代测序read的纠错;
Pacbio测序读长长、通量高、准确度不高,但可通过测序深度弥补,GC偏差低,可进行甲基化的直接测序。
Nanopore测序读长长、通量高、准确度低,不可通过测序深度弥补,但可通过Illumina read 纠错。
随着测序技术的发展和成熟,逐渐形成基因测序产业链
如果你想了解更多企业的话:全球生物数据库与生物软件地图册
常用格式介绍
SRA:NCBI SRA数据库存放格式
SRA(Sequence Read Archive):SRA是一个数据库,NCBI为了解决高通量数据庞大的存储压力而设计的一种数据压缩方案。 一般使用fastq-dump或fasterq-dump来将其转换为fastq格式的数据,才能做后续分析。
FASTQ:高通量数据存储格式
在illumina的测序文件中,采用双端测序(paired-end),一个样本得到的是seq_1.fastq.gz和seq_2.fastq.gz两个文件,每个文件存放一段测序文件。在illumina的测序的cDNA短链被修饰为以下形式(图源见水印):

两端的序列是保护碱基(terminal sequence)、接头序列(adapter)、索引序列(index)、引物结合位点(Primer Binding Site):其中 adapter是和flowcell上的接头互补配对结合的;index是一段特异序列,加入index是为了提高illumina测序仪的使用率,因为同一个泳道可能会测序多个样品,样品间的区分就是通过index区分。参考:illumina 双端测序(pair end)、双端测序中read1和read2的关系。
在illumina公司测得的序列文件经过处理以fastq文件协议存储为*.fastq格式文件。测序得到的段片段称为read,在fastq文件中每4行存储一个read。
第一行:以@开头接ReadID和其他信息 第二行:read测序信息 第三行:规定必须以“+”开头,后面跟着可选的ID标识符和可选的描述内容,如果“+”后面有内容,该内容必须与第一行“@”后的内容相同 第四行:每个碱基的质量得分,与第二行的序列相对应。记分方法是利用ERROR P经过对数和运算分为40个级别分别与ASCII码的第33号!和第73号I对应。用ASCII码表示碱基质量是为了减少文件空间占据和防止移码导致的数据损失。
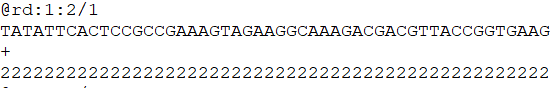
更多可见:我要自学生信之生信基础:FASTA 与 FASTQ
SAM/BAM:高通量数据比对存放格式
SAM文件是由比对产生的以tab建分割的文件格式,BAM是SAM文件的二进制压缩版本。使用samtools view -S -b -o my.bam my.sam可以将SAM文件转换为BAM文件。

BED:基因组浏览器常用格式
BED 文件格式提供了一种灵活的方式来定义的数据行,以用来描述注释信息。BED行有3个必须的列和9个额外可选的列。每行的数据格式要求一致。
必须包含的3列:
(1). chrom - 染色体名字(e.g. chr3,chrY, chr2_random)或scafflold 的名字(e.g. scaffold0671 )。 (2). chromStart - 染色体或scaffold的起始位置,染色体第一个碱基的位置是0。 (3). chromEnd - 染色体或scaffold的结束位置,染色体的末端位置没有包含到显示信息里面。例如,首先得100个碱基的染色体定义为chromStart =0 .chromEnd=100, 碱基的数目是0-99。
9 个额外的可选列: (4). name - 指定BED行的名字,这个名字标签会展示在基因组浏览器中的bed行的左侧。 (5). score - 0到1000的分值,如果在注释数据的设定中将原始基线设置为1,那么这个分值会决定现示灰度水平(数字越大,灰度越高) (6). strand - 定义链的方向,’’+” 或者”-”。 (7). thickStart - 起始位置(The starting position atwhich the feature is drawn thickly)(例如,基因起始编码位置)。 (8). thickEnd - 终止位置(The ending position at whichthe feature is drawn thickly)(例如:基因终止编码位置)。 (9). itemRGB - 是一个RGB值的形式, R, G, B (eg. 255, 0,0), 如果itemRgb设置为’On”, 这个RBG值将决定数据的显示的颜色。 (10). blockCount - BED行中的block数目,也就是外显子数目。 (11). blockSize - 用逗号分割的外显子的大小, 这个item的数目对应于BlockCount的数目。 (12). blockStarts - 用逗号分割的列表, 所有外显子的起始位置,数目也与blockCount数目对应。
GFF/GTF
GFF(General Feature Format)是基于Sanger GFF2的一种格式。GFF有9个必需字段,这些字段必须用制表符分隔。如果字段用空格而不是制表符分隔,则将不能正确显示。GTF (Gene Transfer Format, GTF2.2)是GFF的一种扩展格式。


转录组分析
分析流程
转录组学分析,是基于RNA反转录形成cDNA文库,有两个重要的内容, 即为对转录本表达的丰度和新的转录本的检测。根据有无参考基因可以分为有参转录组分析和无参转录组分析。
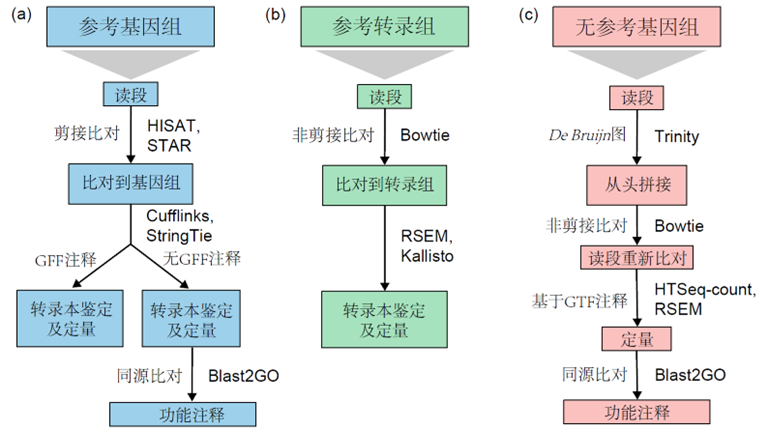
(a)有参,需要分析新转录本 在可以获得物种参考基因组的情况下,如果需要研究可变剪切事件和识别新转录本,则应将reads比对到参考基因组上。
这也是有参物种做转录组最常用的分析模式,其分析步骤如下:
- Reads与基因组比对:TopHat\HISAT2\STAR
- 基于比对信息组装转录本:Cufflinks\Stringtie
- 与基因组注释GFF文件进行比较,鉴定馨转录本
- 利用Blast2GO等工具对新转录本进行功能注释
- 基因或转录本表达定量
- 差异分析和功能富集分析
(b)有参,只分析已知转录本 可以获得物种全转录组的情况下,比如人,小鼠,拟南芥等模式生物,同时实验目的很明确,就是分析已知转录本的表达,那就可以直接基于基因组注释信息中提取出的转录本序列来进行后续分析。该分析模式分析流程简单、速度快,其具体分析步骤如下:
- Reads与转录本序列进行比对:Bowtie
- 转录本表达定量:RSEM\Kallisto等进行定量
- 差异分析和功能富集分析
(c)无参考基因组的转录组 而对于没有参考基因组的物种,或者基因组组装不好的物种,可以使用从头组装的方法处理RNA-seq数据,即先使用测序数据组装一套转录本,再基于转录本进行后续分析。其分析步骤如下:
Reads De novo组装转录本序列
①Trinity软件拼接出transcript,选取其中最长的作为unigene,以N50、N90评价拼接效果。Unigene作为ref用于后续分析。
②Corset软件,在Trinity拼接基础上,根据转录本间Shared Reads将转录本聚合为许多cluster,再结合不同样本间的转录本表达水平及H-Cluster算法,将样本间有表达差异的转录本从原cluster分离,建立新的cluster,最终每个cluster被定义为“Gene”。该方法聚合冗余转录本,并提高差异表达基因的检出率。
Reads 回比组装好的转录本序列:Bowtie
转录本表达定量:htseq-count
利用Blast2GO等工具对转录本进行功能注释,作为Unigene参考注释信息
差异表达分析和功能分析
参考:转录组分析流程 无参转录组 UniGene 无参转录组流程+代码
图源:Conesea et al. ,2016
流程比较
1)tophat2 + cufflink + cuffdiff
参考:流程+代码
初代流程,现在基本不用了
- Star的比对速度是tophat的50倍,hisat更是star的1.2倍。 stringTie的组装速度是cufflinks的25倍,但是内存消耗却不到其一半。 Ballgown在差异分析方面比cuffdiff更高的特异性及准确性,且时间消耗不到cuffdiff的千分之一 Bowtie2+eXpress做质量控制优于tophat2+cufflinks和bowtie2+RSEM Sailfish更是跳过了比对的步骤,直接进行kmer计数来做QC,特异性及准确性都还行,但是速度提高了25倍 kallisto同样不需要比对,速度比sailfish还要提高5倍!!!
2021年nature communications上一篇文章用到的这一流程,但用于早期数据
2)HISAT2+Stringtie+DESeq2
上游分析(数据处理)
上游处理步骤包括质量检测、质量控制、比对、定量。通过质量检测,原始数据的各种问题将会呈现出来,接下来的质量控制就是为了解决原始数据的质量问题。比对是将reads比对到染色体或者基因,并生成sam或者bam文件记录比对情况以及质量。定量既是统计比对到同一位置的reads,当然并不是比对上就计数,还有一些其他的筛选条件,比如比对质量过低或者比对到多个位置的reads就不能用来计数。而免比对的定量软件kallisto和salmon不会生成sam或bam文件而直接进行定量,仅输出定量文件。
参考:上游处理工具介绍
这里我们分析的大肠杆菌注释信息比较详细,进行有参分析,只分析已知转录本。
数据下载
① 基因组和基因注释数据
可以在ncbi\ensemble等下载
Ensembl基因组的不同版本详见README和高通量测序数据处理学习记录(零):NGS分析如何选择合适的参考基因组和注释文件
这里是老师直接给的

② 表达数据
- 自己做实验的就是得到的测序数据
- 如果没有做实验,可以在文章中找到SRR号,然后运行以下命令下载
prefetch SRRxxx
下面的分析基于大肠杆菌的数据,两个时间点5个重复,双端测序
质检
测序结果的好坏会影响后续数据分析的可靠性,故在测序完成后需要通过一些指标对原始测序结果进行评估,如GC含量、序列重复程度、是否存在接头等。如果有问题就需要进行质控。如果经过处理后的测序结果评估仍然较差,说明该样本的测序质量较低,应慎重考虑是否用于后续数据分析。
fastqc
输入:原始测序数据
输出:multiqc_report.html
# 数据储存在~/project/rna.coli/data
dir=~/project/rna.coli
# 原始数据评估 0m19.530s
mkdir qc
time fastqc -o ${dir}/qc ${dir}/data/*.fq -t 5
#合并
time multiqc ${dir}/qc -o ${dir}/qc -n multiqc_rawdata.html
参数设置
- Usage: multiqc [OPTIONS]
-n/--name# 更改输出文件的名称,默认输出文件名:multiqc_data、multiqc_report.html-o/-outdir:在指定的输出目录中创建报告 #若数据在当前目录下输入multiqc . 即可--ignore忽略掉某些文件
结果解读:
Per base sequence quality
横轴是测序序列的碱基,纵轴是质量得分,质量值Q = -10*log10(error P),即20表示1%的错误率,30表示0.1%的错误率。图中每1个箱线图(又称盒须图),都是该位置的所有序列的测序质量的一个统计,分别表示最小值、下四分位数(第25百分位数)、中位数(第50百分位数)、上四分位数(第75百分位数)以及最大值,图中蓝色的细线是各个位置的平均值的连线。
Per sequence quality scores
当峰值处于quality为0-20(错误率1%)时,报错。
Per sequence sequence content
- 正常测序数据为频率相近的四种碱基,无位置差异。表现在图上的话,四条线应该是平行且接近。
- 当任意位置A/T与G/C相差大于10%报警告,大于20%报错
Per base GC content
横轴为GC含量,纵轴为read计数。红色为实际测得,蓝色为理论分布。如果曲线形状不符,代表文库污染
Adapter Content
正常情况是趋于0的直线,也就是说序列两端Adapter已经去除干净;如果有Adapter,需要先用cutadapt去接头
fastp
和上一种方法一样,只是这个做了整合,包含了qc\trim等默认参数。用这个就不用前面的trimmomatic了。但感觉现在生态还不稳定。缺点是:不能通过multiqc整合
#双端测序paired end(PE)
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o our.R1.fq.gz -O out.R2.fq.gz
#批处理
#放处理后文件的目录下执行命令
for i in 1 2 3 4 5;do
{
fastp -i ~/project/rna_coli/data/t1_${i}.1.fq -o t1_${i}.1.fq -I ~/project/rna_coli/data/t1_${i}.2.fq -O t1_${i}.2.fq -w 8 --html 1_${i}.html --json 1_${i}.json
fastp -i ~/project/rna_coli/data/t2_${i}.1.fq -o t2_${i}.1.fq -I ~/project/rna_coli/data/t2_${i}.2.fq -O t2_${i}.2.fq -w 8 --html 2_${i}.html --json 2_${i}.json
}
done
参数说明
-w 8设置进程数8,默认2,max=16,或者是–thread 8-z 5可以设置gzip压缩级别,默认4,1~9 1快-9小--json设置输出json文件名--html设置输出html文件名更多请参见:https://www.jianshu.com/p/8fa2ed15dfaa
输出结果
质量值图
GC含量图:杂质或建库不均匀
ATGC比例图:引物的偏好性
质控:去接头和低质量
trimmomatic
这里我们的原始数据可以,不用进行质控,但保证流程的完整性,这里还是跑一下
帮助文档:http://www.usadellab.org/cms/index.php?page=trimmomatic
对于下机的FASTQ数据需要进行质控和预处理,以保证下游分析输入的数据都是干净可靠的。主要进行以下处理
去接头:分为index+adapter 为什么会有接头呢?
文库的插入片段太短,导致片段被测通,还测到了adapter的位置,好像没理解?给你看下我们的测序片段的组成:adapter+index+插入片段+adapter
测序引物的位点仍然在接头(index)的左边,这样就会把index也测到
去掉N多的测序片段
一个测序片段如果有**很多碱基无法识别(就会出现很多的N),说明测序质量不高或者测序时出现了问题,**这个需要注意,即便去掉了N,也要看下是什么原因引起的,不然治标不治本
去质量评分低的片段
去末端一定数量的片段
随着读长的增加,酶活性下降,荧光强度也在下降,因此测序质量下降是自然趋势
inputdir=~/project/rna.coli
mkdir trim_out
# 调用for循环批处理文件
for filename in ${inputdir}/data/*.1.fq
do
# 提取双端公共文件名,并输出检验
base=$(basename $filename .1.fq)
echo $base
# trimmomatic去接头和低质量
trimmomatic PE -phred33 ${inputdir}/data/${base}.1.fq ${inputdir}/data/${base}.2.fq ${inputdir}/trim_out/${base}_forward_paired.fq.gz ${inputdir}/trim_out/${base}_forward_unpaired.fq.gz ${inputdir}/trim_out/${base}_reverse_paired.fq.gz ${inputdir}/trim_out/${base}_reverse_unpaired.fq.gz ILLUMINACLIP:/home/leixingyi/miniconda3/pkgs/trimmomatic-0.39-hdfd78af_2/share/trimmomatic-0.39-2/adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
done
参数说明
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:去接头 这里我因为运行时报错:TruSeq3-PE.fa (没有那个文件或目录),所以给ILLUMINACLIP后面加上了路径
LEADING:3 TRAILING:3:去除前导链与后随链的低质量或N碱基(质量3以下)
SLIDINGWINDOW:4:15:四碱基扫描框,当每碱基的平均质量低于15时切割
MINLEN:36:删掉低于36碱基的reads
质控后再评估
# 质控后数据评估
mkdir qc/qc_trim
fastqc ${inputdir}/trim_out/*.fq.gz -o ${inputdir}/qc/qc_trim/ -t 5
multiqc ${inputdir}/qc/qc_trim -o ${inputdir}/qc -n multiqc_trimdata.html #合并
K-mer过滤
只对高覆盖度中的低丰度kmer剪切(更可能是测序错误);低覆盖度保留
宏基因组公众号教程里面进行的这一步,具体可见:宏基因组 质控+khmer
其它:k-mer算法解释
比对
序列的比对是RNA分析流程中核心的一步。序列比对中的错配,插入、缺失可以识别出样本和基因组之间的多态性,甚至可以找出肿瘤样本中的gene fusion。而map到没有注释的基因可能是新的编码基因,或者是非编码RNA。同时RNA-seq的序列比对可以揭示新的选择性剪接和同工型(isoform)。
此外,序列的比对也可以用作精确定量基因或者转录本的表达量,因为显然表达丰度与产生的reads是直接成比例的(需要标准化)
参考:rna-seq上游
比对策略
比对是指将reads匹配到参考基因组或转录组的相应位置上(有参),根据比对策略不同分为非剪接比对和剪接比对
可变剪接(概念)
又称选择性剪切,是指基因转录后的mRNA前体经过不同的RNA剪切方式连接外显子形成成熟mRNA的过程。使得一个基因可能会产生多条转录本,即异构体(isoform)
研究表明,动态可变剪切方式控制着大脑皮质发育中细胞的分化命运。
最早?1991年Adams等研究报道的就是人脑组织的部分转录组
因为可变剪切,RNA-Seq reads由于不包含内含子,所以来自外显子边界处的 reads被重新 mapping回基因组时,会被中间的内含子分开,这种情况叫做splice-alignment。
策略1:非剪接比对
不考虑可变剪切。当研究对象的注释信息相对完善,或关注已知基因或转录本的表达情况,就可以选择非剪接比对直接将reads匹配到多有转录本异构体上。
常用工具:
- Bowtie:用户手册;Bowtie和一般的比对工具如blast不一样,他适用于短reads比对到大的基因组上,尽管它也支持小的参考序列像amplicons和长达1024的reads。Bowtie采用基因组索引和reads的数据集作为输入文件并输出比对的列表。Bowtie设计思路是,1)短序列在基因组上至少有一处最适匹配, 2)大部分的短序列的质量是比较高,3)短序列在基因组上最适匹配的位置最好只有一处。这些标准基本上和RNA-seq, ChIP-seq以及其它一些正在兴起的测序技术或者再测序技术的要求一致。
- BWA
策略2:剪接比对
考虑到reads匹配的基因区域可能会被内含子隔开而不连续,故在剪接位点附近的reads可能会部分比对到剪接点两侧外显子上。没有比对到的可能是注释不完全。
常用工具:
TopHat:工作原理如下
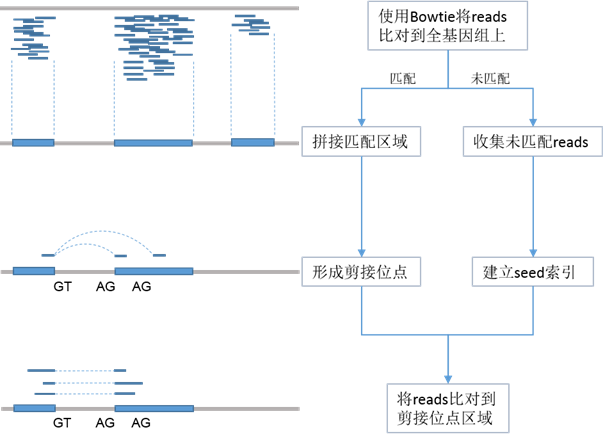
当一个read的几个segmens可以map到基因组时,Tophat推断这个read跨越了剪接位点同时推测剪接位点的位置。通过处理每个‘initially unmappable’ read,Tophat在没有剪接位点注释的信息下能够在转录组上建立起一个剪接位点的索引。
如何推测剪接位点可以参考这篇
关于如何建立索引一直都不清楚,看到这样一个说法觉得比较生动:就类似于像建立图书馆的索引,这样你找书的时候速度就会快很多,仅此而已。
进一步的,可以从算法角度了解索引等:bowtie算法原理探究
STAR:需要更大内存,运行时间也更长
HISAT: 搜先使用Bowtie将reads匹配到全基因组上,成功匹配到reads的区域用于建立潜在的剪接体结构,未能匹配的reads被收集起来建立索引,然后比对到剪接体区域,寻找可能的剪接位点。
2015年发布的HISAT是TopHat的优化版,应用了两类不同的索引类型:代表全基因组的全局FM索引和大量的局部小索引
有这么一种说法:使用BWA来进行ChIP-seq测序,使用bowtie来map DNA测序,使用tophat来map RNA测序。
此外还有salmon这类align-free工具
hisat2
输入:
- 基因组文件
输出:
${outdir}/${base}.sorted.bam: 比对后按染色体位置排序${outdir}/${base}.sorted.bam.bai:所有的排序文件建立的索引
1)建立索引
常见物种可以直接在官网下载索引,截止2022年5月6号,已有的索引如下:

这里没有大肠杆菌的,所以我们自己建立
# 建立基因组索引 0m2.248s
mkdir hisat2
time hisat2-build -p 5 $inputdir/data/genome.fa $inputdir/hisat2/genome.index
---
# 因为有gtf文件所以尝试做gtf和genome综合索引,但得到的genome.ss文件是空的,可能是因为注释文件里没有相关信息
# 提取外显子和可变剪切信息
extract_exons.py $inputdir/data/genome.gtf > $inputdir/hisat2/genome.exon
extract_splice_sites.py $inputdir/data/genome.gtf > $inputdir/hisat2/genome.ss
# 建立索引
hisat2-build $inputdir/data/genome.fa --exon $inputdir/hisat2/genome.exon --ss $inputdir/hisat2/genome.ss -p 5 coli
-p:线程
genome.fa:输入基因组文件
coli.index:输出索引。输出8个索引
2)比对统计
# 双端
# 定义输入输出文件夹
index=~/project/rna.coli/hisat2/genome.index
inputdir=~/project/rna.coli/data
outdir=~/project/rna.coli/hisat2
# 调用for循环批处理文件
for filename in ${inputdir}/*.1.fq
do
# 提取双端公共文件名,并输出检验
base=$(basename $filename .1.fq)
echo $base
# 比对并转化
hisat2 -p 16 -x ${index} -1 ${inputdir}/${base}.1.fq -2 ${inputdir}/${base}.2.fq -S ${outdir}/${base}.align.sam
done
--dta报告,如果后续用Stringtie处理一定要加上这个-f输入文件为FASTA格式-x参考基因组索引文件的前缀。-1双端测序结果的第一个文件。-2双端测序结果的第二个文件。–sra-acc输入SRA登录号,比如SRR353653,多组数据之间使用逗号分隔。HISAT将自动下载并识别数据类型,进行比对。-S指定输出的SAM文件。
3)sam文件处理
使用samtools对sam文件排序并转换为bam文件。.sam文件的二进制形式,相当于压缩
有两种排序方式:
-n:按照read名称进行排序-p:按染色体?
# 双端
# 定义输入输出文件夹
index=~/project/rna.coli/hisat2/genome.index
inputdir=~/project/rna.coli/data
outdir=~/project/rna.coli/hisat2
# 调用for循环批处理文件
for filename in ${inputdir}/*.1.fq
do
# 提取双端公共文件名,并输出检验
base=$(basename $filename .1.fq)
echo $base
# 比对并转化
hisat2 -p 16 -x ${index} -1 ${inputdir}/${base}.1.fq -2 ${inputdir}/${base}.2.fq -S ${outdir}/${base}.align.sam
samtools view -S ${outdir}/${base}.align.sam -b > ${outdir}/${base}.align.bam
samtools sort ${outdir}/${base}.align.bam -o ${outdir}/${base}.sorted.bam
samtools index ${outdir}/${base}.sorted.bam
done
view.sam转化为.bam
sort:排序
index:建立索引,并输出为bai文件,更方便samtool的更多了解可以看 官方文档 以及Nickier老师的samtools的安装和使用
运行samtool时报错:samtools: error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared object file: No such file or directory
cd ~/miniconda3/lib
# 查看链接 得到:libcrypto.so -> libcrypto.so.1.1
ll libcrypto.so
# 建立软链接
ln -s libcrypto.so.1.1 libcrypto.so.1.0.0
# 测试
samtools --help
# 可以用了,但我只能在/lib路径下调用samtools。有时间再来研究什么原因
4)比对结果可视化
.bam文件可以使用基因组浏览器IGV( Integrative Genomics Viewer); Genome Maps等可视化,且需要输入index文件和基因组文件
比对结果分析:
- reads匹配百分比可以用来评估总测序精确度和DNA污染程度
- reads分布:分布均匀说明打断随机性较好
- 匹配reads的GC含量:与PCR偏差有关
5)比对结果质量评估
在A survey of best practices for RNA-seq data analysis里面,提到了人类基因组应该有70%~90%的比对率,并且多比对read(multi-mapping reads)数量要少。另外比对在外显子和所比对链(uniformity of read coverage on exons and the mapped strand)的覆盖度要保持一致。 常用工具有
Qualimap http://qualimap.bioinfo.cipf.es/ 依赖Java和R
或者samtools falgstate
samtools flagstate seq_sorted.bam > seq_sorted.flagstate
-g# 注释文件中提取对Meta-feature 默认是gene_id-t# 提取注释文件中的Meta-feature 默认是 exon-p#参数是针对paired-end 数据-a#输入GTF/GFF 注释文件-o#输出文件
Tophat
1)bowtie2建索引
mkdir tophat
nohup bowtie2-build ${dir}/data/genome.fa ${dir}/tophat/coli_index >bowtie2-build.log 2>&1 &
2)比对
#-G gtf文件;-o bowtie建立参考基因组索引的公共名(前缀);
nohup tophat2 -p 6 -G ${dir}/data/genome.gtf -o ${dir}/tophat/${base} -p 6 ${dir}/tophat/coli_index ${dir}/data/${base}.1.fq ${dir}/data/${base}.2.fq >tophat.log 2>&1 &
参数:
- -o 输出文件夹
- coli_index :bowtie2建立的索引
比对结果:
拼接
如果仅仅关注已知基因表达情况,不需要鉴定新转录本可以跳过此步骤
stringtie
1)有参转录本拼接
2)转录本整合
3)转录本注释文件比较
使用gffcompare对整合后转录本注释文件与参考注释比较,获得可能的新转录本信息
Cufflinks
计算基因表达量的另一个问题是,因为选择性剪接的原因,几个不同的转录本(isoform)可能拥有相同的外显子,此时难以确定reads到底来自其中哪个转录本(isoform)。所以能否确定所有的splice variants(isoform)决定着表达量计算的准确性。而这个又很难确定,所以cufflinks通过map到基因组的reads组装起一个简陋的转录组,用reads拼接成含有重叠部分但是长度不同的转录本(称作”transfrags“,作为splice variants的推测。拼接以后,Cufflinks计算使用严格的统计模型来计算每个transfrag的表达量。
当有多个样本的时候,一种方法是将所有样本的reads合起来,拼接成一个转录组。这种方法的缺点是:
- 大量reads带来的计算不便,需要更高配置的服务器和大量的时间(可以参考 de no vo Trinity,动辄上百G的内存需求)
- 多个样本重叠使得确定所有的splice variants(isoform)更加困难
所以Cufflinks采用的策略是先单独拼接每一个样本的reads,然后使用cuffmerge来综合所有样本的拼接结果
cufflinks套装有很多,我们主要使用的只有三个
Cufflinks是用来处理tophat的输出的bam文件然后输出gtf文件
cuffmerge把多个样本的gtf文件合并的,也没啥子用,主要是测多个样本可能会需要
cuffdiff算出分组的bam文件里面的差异基因。主要是发现转录本表达,剪接,启动子使用的明显变化。
cufflinks和cuffmerge和cuffdiff需要的文件各不相同
对cufflinks来说,需要的是tophat输出的bam文件
对cuffmerge来说,需要的是多次运行cufflinks输出的多个gtf文件
对cuffdiff来说,需要的多个样本的tophat的bam文件
参考:Tophat+cufflinks 流程+代码 tophat+cufflink 代码
# 定义输入输出文件夹
index=~/project/rna.coli/hisat2/genome.index
indir=~/project/rna.coli/hisat2
# 调用for循环批处理文件
for filename in ${inputdir}/*.1.fq
do
# 提取双端公共文件名,并输出检验
base=$(basename $filename .1.fq)
echo $base
# Cufflinks,分别拼接每一个样本的转录组
mkdir -p cufflink/cuffmerge
mkdir -p cufflink/cuffdiff
time cufflinks -G ${dir}/data/genome.gtf -o ${dir}/cufflink/${base} -p 6 ${dir}/tophat/${base}/accepted_hits.bam
#运行cuffmerge,将所有样本拼接好的转录组合成一个
#参数 -g 添加基因组注释文件,可选
#参数 -s 为参考基因组 fasta格式
echo ${dir}/cufflink/${base}/transcripts.gtf >> ${dir}/cufflink/assemblies.txt
time cuffmerge -g ${dir}/data/genome.gtf -s ${dir}/data/genome.fa -o ${dir}/cufflink/cuffmerge -p 6 ${dir}/cufflink/assemblies.txt
done
#运行cuffdiff(没想到整合方法)
#-b 参考基因组mm10的基因组序列
#-u 对多比对位置的read加权然后分配比对到基因组上
#-L 样本名
time cuffdiff -o ${dir}/data/cufflink/cuffdiff -b ${dir}/data/genome.fa -p 16 -L t1,t2 -u ${dir}/cufflink/cuffmerge/merged.gtf \
${dir}/tophat/t1_1/accepted_hits.bam,${dir}/tophat/t1_2/accepted_hits.bam,${dir}/tophat/t1_3/accepted_hits.bam,${dir}/tophat/t1_4/accepted_hits.bam,${dir}/tophat/t1_5/accepted_hits.bam \
${dir}/tophat/t2_1/accepted_hits.bam,${dir}/tophat/t2_2/accepted_hits.bam,${dir}/tophat/t2_3/accepted_hits.bam,${dir}/tophat/t2_4/accepted_hits.bam,${dir}/tophat/t2_5/accepted_hits.bam
cufflinks
- -G | –GTF 提供一个GFF文件,以此来计算isoform的表达。此时,将不会组装新的transcripts, 程序会忽略和reference transcript不兼容的比对结果
- -g | –GTF-guide 提供GFF文件,以此来指导转录子组装(RABT assembly)。此时,输出结果会包含reference transcripts和novel genes and isforms。
cuffidff
- -b | –frag-bias-correct(一般是genome.fa) 提供一个fasta文件来指导Cufflinks运行新的bias detection and correction algorithm。这样能明显提高转录子丰度计算的精确性。
- -L | –lables default: q1,q2,…qN 给每个sample一个样品名或者一个环境条件一个lable
- -u | –multi-read-correct 让Cufflinks来做initial estimation步骤,从而更精确衡量比对到genome多个位点 的reads。
更多见:Cufflinks参数说明
Cufflinks输出结果
- transcripts.gtf
该文件包含Cufflinks的组装结果isoforms。前7列为标准的GTF格式,最后一列为attributes。其每一列的意义:

ispforms.fpkm_tracking
isoforms(可以理解为gene的各个外显子)的fpkm计算结果
genes.fpkm_tracking
gene的fpkm计算结果
cuffdiff输出结果:
genes_exp.diff文件后续分析
Trinity
运行该软件时,主要分三部分运行,依次为Inchworm、Chrysalis和Butterfly

运行Inchworm时,其目的是将每条短读长的reads打断成固定长度的Kmer(默认为25 bp)形成一个Kmer库,再将所有Kmer按其出现的次数进行排序,选择出现次数最高的Kmer进行3端延伸一个碱基(A、T、G、C),延伸后统计靠近3端25bp的Kmer在库中出现的次数,选择次数最高的那条路径(如有多条路径出现的次数一样,则几条路径保留),继续向3端延伸至不能延伸为止。同理进行5端延伸,最终形成一定长度的Contigs。
每形成一个Contigs时,将形成该Contigs的kmer从Kmer库中去除掉,继续选择次数最高的Kmer进行延伸。最终用完Kmer库中的Kmer,将Contigs放在一起形成一个contigs库。运行Chrysalis时,对Contigs库中的Contigs按照一定条件进行聚类,之后对每类Contigs构建de bruijin graphs。
最后运行Butterfly,解析上一步构建的de bruijin graphs,形成转录本。将所有转录本输出到一个文件,便是最Trinity终组装的文件。
定量与转录本组装
reads计数
bam文件只有序列比对的情况,而注释信息会标注哪里是基因。比如bam文件里面标注了比对到哪里,注释信息会告诉你这块是什么基因,如何就可以计算多少条reads比对到了该基因。两种策略:
只选择唯一匹配reads计数。这种方式会将多重比对的reads舍弃。
工具如:HTSeq-count; featureCount
保留多重匹配的reads。利用统计模型将多重比对的reads定位到对应的转录本异构体上
工具如:Cufflinks; StringTie; RSEM
FeatureCount
# featurecount通过subread安装
conda install -y subread
# 定义输入输出文件夹
mkdir fea.count
gtf=~/project/rna.coli/data/genome.gtf
inputdir=~/project/rna.coli/hisat2
outdir=~/project/rna.coli/fea.count
# 计数
featureCounts -T 16 -p -t exon -g gene_id -a $gtf -o $outdir/out.count.txt $inputdir/*.sorted.bam >$outdir/count.log 2>&1 &
# 可视化
multiqc out.count.txt.summary
# 提取counts.txt的第1(id)和第7-17列(length) #删除中间多列还是perl更简单!
cat out.count.txt | perl -lane 'splice @F,1,5; print join " ",@F' >featureCounts.txt
-p双端测序,统计fragment而不统计read
-t设置feature-type,-t指定的必须是gtf中有的feature,同时read只有落到这些feature上才会被统计到,默认是“exon”
-g选择gtf中提供的id identifier!!,默认为gene_id
-a参考文件gtf文件名
-0为一系列输入文件
count.log 2>&1 &将标准错误重定向到count.log文件中
&放在命令到结尾,表示后台运行会输出两个文件,out.count.txt记录了定量结果,summary文件统计了定量情况
表达量标准化
reads数受到基因长度、测序深度和测序误差等影响,需要归一化处理后才能用于差异表达分析
- 测序深度:同一条件下,测序深度越深,基因表达的read读数越多。别人1个G,你10个G的数据,reads数量本身就高了。
- 基因长度:同一条件下,基因越长,该基因的read读数越高。越长,打断出的片段就越多
常用的数据标准化方法如FPKM/RPKM/TPM,是一种定量方式,一般可比,但也会有protocol特异的偏好。一般不用于差异基因检测。
$$FPKM= \frac{counts}{\frac{mapped,reads}{10^3}\times\frac{exon,length}{10^6}}=\frac{counts\times10^9}{mapped,reads\times exon,length}$$
式中:
- counts:匹配到该基因区域的reads数量
- mapped reads:该基因长度
- exon length:该样本总reads。使用总reads数矫正测序深度差异,使不同处理组得到的表达量值恒定。
FPKM和TPM是RPKM的衍生方法
FPKM
计算FPKM的三要素:原始counts矩阵,样本总reads数,基因长度。
RPKM与FPKM的区别:一个是read,一个是fragment;RPKM值适用于单末端RNA-seq实验数据,FPKM适用于双末端RNA-seq测序数据。
基于stringtie
string -e -p 2 -G
-e:只列出已知转录本丰度
-p:线程数
-G:注释文件。不关注新转录本可以直接使用参考注释文件
-A:输出基因水平表达风度文件
-o:输出转录本水平表达丰度文件每个bam文件经过计算输出两个GTF注释文件,包括基因水平和转录本水平,且计算了每个基因或转录本的表达丰度FPKM值。提取每个GTF文件中的FPKM值组成最终的基因表达数据矩阵。
基于R
相关的包如countToFPKM,相关信息:https://github.com/AAlhendi1707/countToFPKM
复习一下公式:$FPKM= \frac{counts}{\frac{mapped,reads}{10^3}\times\frac{exon,length}{10^6}}=\frac{counts\times10^9}{mapped,reads\times exon,length}$,其中最难算的是exon length,下面我是通过计算非冗余外显子(exon)长度之和得到的
# R环境下运行
# environment
if (!require("DEseq2"))BiocManager::install("GenomicFeatures")
# 构建txdb对象
library(GenomicFeatures)
rm(list=ls())
txdb = makeTxDbFromGFF('genome.gtf',format = 'gtf')#注释文件
# 计算非冗余外显子(exon)长度之和
# 通过exonsBy获取每个gene上的所有外显子的起始位点和终止位点,然后用reduce去除掉重叠冗余的部分,最后计算长度
exons_gene = exonsBy(txdb,by='gene')
exons_gene_lens = lapply(exons_gene,function(x){sum(width(reduce(x)))})
# 导入计数数据
rawcounts = read.table('featureCounts.txt',header = T)#计数结果,得出方法见上
# 数据整理
rowname = rawcounts[,1]
rawcounts = data.frame(rawcounts[,-1],stringsAsFactors = F)
colnames(rawcounts) = c(paste0('t1_',1:5),paste0('t2_',1:5))
row.names(rawcounts) = rowname
# 检查数据没有出错
str(rawcounts)
# 计算
fpkm = array(0,dim=c(length(rownames(rawcounts)),length(colnames(rawcounts))))
for (i in 1:length(rownames(rawcounts))) {
for (j in 1:length(colnames(rawcounts))) {
fpkm[i,j] = rawcounts[i,j]/as.integer(exons_gene_lens[names(exons_gene_lens)==rownames(rawcounts)[i]])/sum(rawcounts[,j])*10^9
}
}
# 数据整理
colnames(fpkm) = c(paste0('fpkm_t1_',1:5),paste0('fpkm_t2_',1:5))
rownames(fpkm) = rownames(rawcounts)
#fpkm = cbind(fpkm,rawcounts) 看想不想合并
# 输出
write.csv(fpkm,'fpkm.csv')
其它转换方法:没用任何包、用GenomicFeatures自带转化函数
String tie
gtf结果文件中有coverage,TPM和FPKM。此外RSEM也可计算FPKM值。
cd ../bam
ls *bam|while read id;do stringtie $id -p 4 -G /mnt/data/hwb/database/mouse_genome_esml_84/grcm38_tran/Mus_musculus.GRCm38.84.chr.gtf -o ../out_gtf/$(basename $id "bam")gtf;done
下游分析(功能分析)
差异表达分析
- cummeRbund主要是下游可视化分析,基于cufflink,cuffdiff的结果;
- DESeq2,edgeR是基于count的分析结果;
- limma最早是处理芯片数据的,后来有一个limma voom可以处理RNA-Seq数据;
基因表达具有时空特异性,即在不同组织细胞、不同时间下会出现差异。在不同实验条件处理下,基因表达也会有不同的相应模式。差异表达分析的目的是找出不同条件下表达上调、下调或保持稳定的基因,探究实验条件如何影响基因表达并调控相关生物学过程的机制。
常用工具:
- DEseq2针对有生物学重复的样本。(一般情况下应该是都需要生物学重复的)
- edgeR对于单个样本是比较好的。
这两个软件的核心思想是表达量居中的基因或转录本在所有样本中的表达量都应该是相似的,基于分布的标准化方式。都自带归一化算法(标准化),所以导入的数据是原始数据。想了解上面使用偏向也可以从他们的标准化方法入手。
cummeRbund
输入:cuffdiff的所有输出文件所在文件夹
DEseq2
输入:featureCounts.txt:FeatureCount计量结果
输出:DESeq2_diffExpression.csv:差异表达基因
参数设置
- 筛选条件:$\lvert log2FC\rvert$ > 1,padjust < 0.05
# environment
# DEseq2
# install
if (!require("DEseq2"))BiocManager::install("DESeq2")
# data
library(DESeq2)
rm(list=ls())
countData <- read.table('featureCounts.txt',header = T,row.names=1) #未标准化的数据
colnames(countData) = c(paste0('t1_',1:5),paste0('t2_',1:5))
condition<- factor(c(rep('t1',5),rep('t2',5)))
colData <- data.frame(row.names=colnames(countData), condition)
# 构建dds矩阵
dds <- DESeqDataSetFromMatrix(countData, DataFrame(condition), design= ~ condition )
head(dds)
# 差异表达分析
dds <- DESeq(dds)
res <- results(dds) #差异分析结果
#接下来,我们要查看t1 VS t2 的总体结果,并根据p-value进行重新排序。
#利用summary命令统计显示一共多少个genes上调和下调(FDR0.1)
res <- results(dds, contrast=c("condition", "t1", "t2"))##或者res= results(dds)
res <- res[order(res$pvalue),]
summary(res)
#所有结果先进行输出
write.csv(res,file="DESeq2_results.csv")
#获取padj(p值经过多重校验校正后的值)小于0.05,表达倍数取以2为对数后大于1或者小于-1的差异表达基因。
diff_gene_deseq2 <-subset(res, padj < 0.05 & abs(log2FoldChange) > 1) #abs表示绝对值
##或者diff_gene_deseq2 <-subset(res,padj < 0.05 & (log2FoldChange > 1 | log2FoldChange < -1))
dim(diff_gene_deseq2)
write.csv(diff_gene_deseq2,file= "DESeq2_diffExpression.csv")
疑问1:对于重复样本,DESeq分析前要取均值吗?既然输入了colData信息,是不是代表它会自动处理重复
edgeR
if (!require("edgeR"))BiocManager::install("edgeR")
可视化
#火山图
#热图
#veen
# data
rm(list=ls())
DEG = na.omit(read.csv("DESeq2_results.csv",row.names = 1))
##### 火山图 ######
## 使用基础函数plot绘图
plot(DEG$log2FoldChange,-log2(DEG$padj))
## 按照差异基因的log2FoldChange大于1,确定上下调表达基因。
DEG$change = as.factor(ifelse(DEG$padj < 0.05 & abs(DEG$log2FoldChange) > 1,
ifelse(DEG$log2FoldChange > 1 ,'UP','DOWN'),'STABLE'))
# 绘图
library(ggplot2)
library(ggrepel)
library(export)
## 标题
this_tile <- paste0('Cutoff for lnFC is 1',
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',]))
this_tile
## 绘图的时候必须是dataframe,所以需要转换一下。
DEG <- data.frame(DEG)
## 颜色与分组一一对应
Volcano <- ggplot(data=DEG, aes(x=log2FoldChange, y=-log10(padj),color=change)) +
geom_point(shape = 16, size=2) +
theme_set(theme_set(theme_bw(base_size=20))) +
xlab("log2 fold change") +
ylab("-log10 p-value") +
ggtitle( this_tile ) +
theme(plot.title = element_text(size=15,hjust = 0.5)) +
theme_classic()+
scale_colour_manual(values = c('blue','black','red'))
Volcano
##### 热图 #####
# 导入原始计数数据
countData <- read.table('featureCounts.txt',header = T,row.names=1)
colnames(countData) = c(paste0('t1_',1:5),paste0('t2_',1:5))
# 筛选
## 提取差异倍数
library(magrittr)
FC <- DEG$log2FoldChange %>% subset(.,abs(.)>1)
names(FC) <- rownames(subset(DEG,abs(log2FoldChange) > 1))
## 排序差异倍数,提取前100和后100的基因名
## 或者全部的基因名字
DEG_200 <- c(names(head(sort(FC),100)),names(tail(sort(FC),100)))
DEG_all <- c(names(sort(FC)))
## 筛选
dat = countData[DEG_all,]
# 标准化
## 方法1 去中心+标准化
scale_dis <- t(scale(t(dat)))
View(scale_dis)
# 方法2 泊松距离(Poisson Distance)
library(PoiClaClu)
poisd_dis <- PoissonDistance(countData)
## 方法3 欧式距离
library(vegan)
Eucl_dis <- dist(t(array(countData)))
Eucl_dis
# Eucl_dis=dist(countData,method = "euclidean") 或者使用默认函数
# 绘图
library(pheatmap)
group = factor(c(rep('t1',5),rep('t2',5)))
group = factor(c(rep('t1',5),rep('t2',5))) %>% data.frame(row.names=colnames(countData), .)
heatmap <- pheatmap(scale_dis,show_colnames =T,show_rownames = T, cluster_cols = F,border=T)
heatmap
##### veen #####
# 均值
dat.fram <- data.frame(dat)
dat.fram$t1 <-round((dat$t1_1 + dat$t1_2 + dat$t1_3 +dat$t1_4 + dat$t1_5)/5)
dat.fram$t2 <-round((dat$t2_1 + dat$t2_2 + dat$t2_3 +dat$t2_4 + dat$t2_5)/5)
dat2 <- subset(dat.fram, select=c(t1,t2))
# 绘图
library(limma)
venn = vennDiagram(dat2, include = 'both', names = c('t1', 't2'), cex = 2,
counts.col = 'black', circle.col = c('orange', 'purple'))
# export可以将图片保存为pptx,便于修改
graph2ppt(Volcano,file="Volcano_Plot.ppt", width=10, aspectr=1)
graph2ppt(heatmap,file="pheatmap_cluster.ppt", width=10, aspectr=1)
graph2ppt(venn,file="venn.ppt", width=10, aspectr=1)
求均值这里:可以用tidyverse里的group_by函数分组,然后summarise计算均值,不过数据得要转一下,还得加个group,这里数据比较少就直接手动了
差异表达与limma的一些注意事项:https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247490865&idx=1&sn=7b7c6d03ec13a10f4c4f08b756de3695&scene=21#wechat_redirect
聚类分析
富集分析
对差异显著的基因进行GO和KEGG富集,探究相关生物学功能和通路。
共表达网络
WGCNA全称加权基因共表达网络分析 (Weighted correlation network analysis),它是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集, 并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
WGCNA基于两个假设:(1)相似表达模式的基因可能存在共调控、功能相关或处于同一通路,(2)基因网络符合无标度分布。基于这两点,可以将基因网络根据表达相似性划分为不同的模块进而找出枢纽基因。
未完待续
#共表达网络 WGCNA
#GO/KEGG
主要就是id转换。
在线网站:
#蛋白互作网络
string+cytoscape
实验验证
用qPCR进行实验验证RNA-seq的可靠性,目前还没有人会把RNA-seq作为最可靠的证据,一般都是要进行qPCR实验验证的,而且在实验验证时只有十几个基因,可以使用多个重复,结果更加准确。
总结
上游分析主要就是对原始数据进行一些修修剪剪,整理一下给到下游,个性化分析就主要集中在下游了。分析内容有很多。这次跑完,也确实感觉到了,R语言更多的是一个数据处理软件,只要数据处理好了,绘图就只是一条命令了。
还需要继续学习的点:各种方法的原理和算法;R里面的一些命令可以优化;
all right reserved @ 雷心怡 20220501
由于本人能力有限,编写过程中难免存在不足和错漏,恳请大家不吝赐教,以便及时改正。邮箱:2207997703@qq.com。
其它教程可供参考:
①Tophat –> Cufflink –> Cuffdiff
②Subread -> featureCounts -> DESeq2
立小里 笔记 内容不多,但很规整